












For more information, visit http://www.nhlbi.nih.gov/health/health-topics/topics/sca/
What Is Sickle Cell Anemia?
Sickle cell anemia (uh-NEE-me-uh) is the most common form of sickle cell disease (SCD). SCD is a serious disorder in which the body makes sickle-shaped red blood cells. “Sickle-shaped” means that the red blood cells are shaped like a crescent.
Normal red blood cells are disc-shaped and look like doughnuts without holes in the center. They move easily through your blood vessels. Red blood cells contain an iron-rich protein called hemoglobin (HEE-muh-glow-bin). This protein carries oxygen from the lungs to the rest of the body.
Sickle cells contain abnormal hemoglobin called sickle hemoglobin or
Sickle cells are stiff and sticky. They tend to block blood flow in the blood vessels of the limbs and organs. Blocked blood flow can cause pain, serious infections, and organ damage.
Normal Red Blood Cells and Sickle Cells
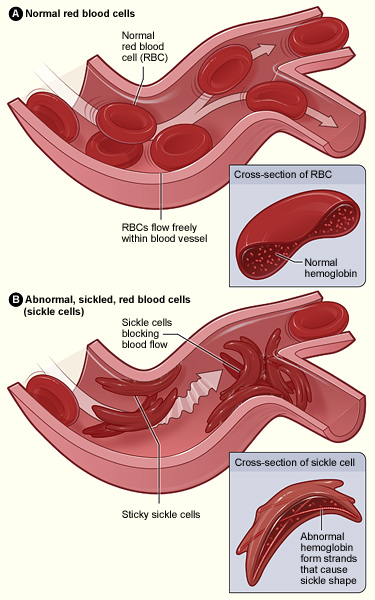
Figure A shows normal red blood cells flowing freely in a blood vessel. The inset image shows a cross-section of a normal red blood cell with normal hemoglobin. Figure B shows abnormal, sickled red blood cells blocking blood flow in a blood vessel. The inset image shows a cross-section of a sickle cell with abnormal (sickle) hemoglobin forming abnormal strands.
Overview
Sickle cell anemia is one type of anemia. Anemia is a condition in which your blood has a lower than normal number of red blood cells. This condition also can occur if your red blood cells don't contain enough hemoglobin.
Red blood cells are made in the spongy marrow inside the large bones of the body. Bone marrow is always making new red blood cells to replace old ones. Normal red blood cells live about 120 days in the bloodstream and then die. They carry oxygen and remove carbon dioxide (a waste product) from your body.
In sickle cell anemia, the number of red blood cells is low because sickle cells don't last very long. Sickle cells usually die after only about 10 to 20 days. The bone marrow can't make new red blood cells fast enough to replace the dying ones.
Sickle cell anemia is an inherited, lifelong disease. People who have the disease are born with it. They inherit two genes for sickle hemoglobin—one from each parent.
People who inherit a sickle hemoglobin gene from one parent and a normal gene from the other parent have a condition called sickle cell trait.
Sickle cell trait is different than sickle cell anemia. People who have sickle cell trait don't have the disease, but they have one of the genes that cause it. Like people who have sickle cell anemia, people who have sickle cell trait can pass the sickle hemoglobin gene on to their children.
Outlook
Sickle cell anemia has no widely available cure. However, treatments can help with the symptoms and complications of the disease. Blood and marrow stem cell transplants may offer a cure for a small number of people.
Over the past 100 years, doctors have learned a great deal about sickle cell anemia. They know its causes, how it affects the body, and how to treat many of its complications.
Sickle cell anemia varies from person to person. Some people who have the disease have chronic (long-term) pain or fatigue (tiredness). However, with proper care and treatment, many people who have the disease can have improved quality of life and reasonable health much of the time.
Due to improved treatments and care, people who have sickle cell anemia are now living into their forties or fifties, or longer.
Other Names for Sickle Cell Anemia
- HbS disease
- Hemoglobin S disease
- Hemoglobin SS disease
- Sickle cell disease (a broad term that includes sickle cell anemia)
- Sickle cell disorders (a broad group of conditions that includes sickle cell anemia)
- Sickling disorder due to hemoglobin S
What Causes Sickle Cell Anemia?
Sickle cell anemia is an inherited disease. People who have the disease inherit two genes for sickle hemoglobin—one from each parent.
Sickle hemoglobin causes red blood cells to develop a sickle, or crescent, shape. Sickle cells are stiff and sticky. They tend to block blood flow in the blood vessels of the limbs and organs. Blocked blood flow can cause pain, serious infections, and organ damage.
Sickle Cell Trait
People who inherit a sickle hemoglobin gene from one parent and a normal gene from the other parent have a condition called sickle cell trait. Their bodies make sickle hemoglobin and normal hemoglobin.
People who have sickle cell trait usually have few, if any, symptoms and lead normal lives. However, some people may have medical complications.
People who have sickle cell trait can pass the sickle hemoglobin gene to their children. The following image shows an example of an inheritance pattern for sickle cell trait.
Example of an Inheritance Pattern for Sickle Cell Trait
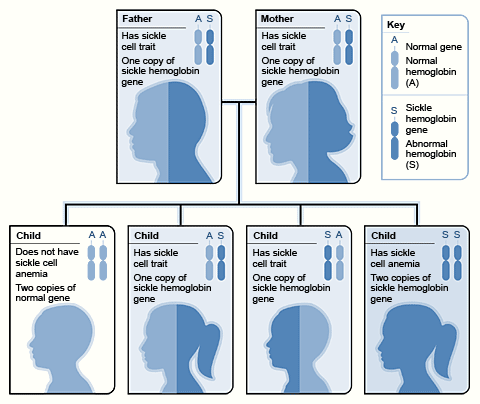
The image shows how sickle hemoglobin genes are inherited. A person inherits two hemoglobin genes—one from each parent. A normal gene will make normal hemoglobin (A). A sickle hemoglobin gene will make abnormal hemoglobin (S).
When both parents have a normal gene and an abnormal gene, each child has a 25 percent chance of inheriting two normal genes; a 50 percent chance of inheriting one normal gene and one abnormal gene; and a 25 percent chance of inheriting two abnormal genes.
Who Is at Risk for Sickle Cell Anemia?
Sickle cell anemia is most common in people whose families come from Africa, South or Central America (especially Panama), Caribbean islands, Mediterranean countries (such as Turkey, Greece, and Italy), India, and Saudi Arabia.
In the United States, it's estimated that sickle cell anemia affects 70,000–100,000 people, mainly African Americans. The disease occurs in about 1 out of every
More than 2 million Americans have sickle cell trait. The condition occurs in about
What Are the Signs and Symptoms of Sickle Cell Anemia?
The signs and symptoms of sickle cell anemia vary. Some people have mild symptoms. Others have very severe symptoms and often are hospitalized for treatment.
Sickle cell anemia is present at birth, but many infants don't show any signs until after 4 months of age.
The most common signs and symptoms are linked to anemia and pain. Other signs and symptoms are linked to the disease's complications.
Signs and Symptoms Related to Anemia
The most common symptom of anemia is fatigue (feeling tired or weak). Other signs and symptoms of anemia may include:
- Shortness of breath
- Dizziness
- Headaches
- Coldness in the hands and feet
- Paler than normal skin or mucous membranes (the tissue that lines your nose, mouth, and other organs and body cavities)
- Jaundice (a yellowish color of the skin or whites of the eyes)
Signs and Symptoms Related to Pain
Sudden pain throughout the body is a common symptom of sickle cell anemia. This pain is called a sickle cell crisis. Sickle cell crises often affect the bones, lungs, abdomen, and joints.
These crises occur when sickled red blood cells block blood flow to the limbs and organs. This can cause pain and organ damage.
The pain from sickle cell anemia can be acute or chronic, but acute pain is more common. Acute pain is sudden and can range from mild to very severe. The pain usually lasts from hours to as long as a week or more.
Many people who have sickle cell anemia also have chronic pain, especially in their bones. Chronic pain often lasts for weeks or months and can be hard to bear and mentally draining. Chronic pain may limit your daily activities.
Almost all people who have sickle cell anemia have painful crises at some point in their lives. Some have these crises less than once a year. Others may have crises once a month or more. Repeated crises can damage the bones, kidneys, lungs, eyes, heart, and liver. This type of damage happens more often in adults than in children.
Many factors can play a role in sickle cell crises. Often, more than one factor is involved and the exact cause isn't known.
You can control some factors. For example, the risk of a sickle cell crisis increases if you're dehydrated (your body doesn't have enough fluids). Drinking plenty of fluids can lower the risk of a painful crisis.
You can't control other factors, such as infections.
Painful crises are the leading cause of emergency room visits and hospital stays for people who have sickle cell anemia.
Complications of Sickle Cell Anemia
Sickle cell crises can affect many parts of the body and cause many complications.
Hand-Foot Syndrome
Sickle cells can block the small blood vessels in the hands and feet in children (usually those younger than 4 years of age). This condition is called hand-foot syndrome. It can lead to pain, swelling, and fever.
Swelling often occurs on the back of the hands and feet and moves into the fingers and toes. One or both hands and/or feet may be affected at the same time.
Splenic Crisis
The spleen is an organ in the abdomen. Normally, it filters out abnormal red blood cells and helps fight infections. In some cases, the spleen may trap red blood cells that should be in the bloodstream. This causes the spleen to grow large and leads to anemia.
If the spleen traps too many red blood cells, you may need blood transfusions until your body can make more cells and recover.
Infections
Both children and adults who have sickle cell anemia may get infections easily and have a hard time fighting them. This is because sickle cell anemia can damage the spleen, an organ that helps fight infections.
Infants and young children who have damaged spleens are more likely to get serious infections that can kill them within hours or days. Bloodstream infections are the most common cause of death in young children who have sickle cell anemia.
Medicines and vaccines can help prevent severe illness and death. For example, vaccines are available for infections such as meningitis, influenza, and hepatitis.
Getting treatment right away for high fevers (which can be a sign of a severe infection) also helps prevent death in infants and children who have sickle cell anemia.
It's also important to get treatment right away for a cough, problems breathing, bone pain, and headaches.
Acute Chest Syndrome
Acute chest syndrome is a life-threatening condition linked to sickle cell anemia. This syndrome is similar to pneumonia. An infection or sickle cells trapped in the lungs cause acute chest syndrome.
People who have this condition often have chest pain, shortness of breath, and fever. They also often have low oxygen levels and abnormal chest x ray results.
Pulmonary Hypertension
Damage to the small blood vessels in the lungs makes it hard for the heart to pump blood through the lungs. This causes blood pressure in the lungs to rise.
Increased blood pressure in the lungs is called pulmonary hypertension (PH). Shortness of breath and fatigue are the main symptoms of PH.
Delayed Growth and Puberty in Children
Children who have sickle cell anemia often grow more slowly than other children. They may reach puberty later. A shortage of red blood cells causes the slow growth rate. Adults who have sickle cell anemia often are slender or smaller in size than other adults.
Stroke
Two forms of stroke can occur in people who have sickle cell anemia. One form occurs if a blood vessel in the brain is damaged and blocked. This type of stroke occurs more often in children than adults. The other form of stroke occurs if a blood vessel in the brain bursts.
Either type of stroke can cause learning problems and/or lasting brain damage, long-term disability, paralysis (an inability to move), or death.
Eye Problems
Sickle cells also can affect the small blood vessels that deliver oxygen-rich blood to the eyes. Sickle cells can block these vessels or cause them to break open and bleed. This can damage the retinas—thin layers of tissue at the back of the eyes. The retinas take the images you see and send them to your brain.
This damage can cause serious problems, including blindness.
Priapism
Males who have sickle cell anemia may have painful, unwanted erections. This condition is called priapism (PRI-a-pizm). It happens because the sickle cells block blood flow out of an erect penis. Over time, priapism can damage the penis and lead to impotence.
Gallstones
When red blood cells die, they release their hemoglobin. The body breaks down this protein into a compound called bilirubin. Too much bilirubin in the body can cause stones to form in the gallbladder, called gallstones.
Gallstones may cause steady pain that lasts for 30 minutes or more in the upper right side of the belly, under the right shoulder, or between the shoulder blades. The pain may happen after eating fatty meals.
People who have gallstones may have nausea (feeling sick to the stomach), vomiting, fever, sweating, chills, clay-colored stools, or jaundice.
Ulcers on the Legs
Sickle cell ulcers (sores) usually begin as small, raised, crusted sores on the lower third of the leg. Leg sores may occur more often in males than in females. These sores usually develop in people who are aged 10 years or older.
The cause of sickle cell ulcers isn't clear. The number of ulcers can vary from one to many. Some heal quickly, but others persist for years or come back after healing.
Multiple Organ Failure
Multiple organ failure is rare, but serious. It happens if you have a sickle cell crisis that causes two out of three major organs (lungs, liver, or kidneys) to fail. Often, multiple organ failure occurs during an unusually severe pain crisis.
Symptoms of this complication are fever, rapid heartbeat, problems breathing, and changes in mental status (such as sudden tiredness or confusion).
How Is Sickle Cell Anemia Diagnosed?
A simple blood test, done at any time during a person's lifespan, can detect whether he or she has sickle hemoglobin. However, early diagnosis is very important.
In the United States, all States mandate testing for sickle cell anemia as part of their newborn screening programs. The test uses blood from the same blood samples used for other routine newborn screening tests. The test can show whether a newborn infant has sickle hemoglobin.
Test results are sent to the doctor who ordered the test and to the baby's primary care doctor. It's important to give the correct contact information to the hospital. This allows the baby's doctor to get the test results as quickly as possible.
Health providers from a newborn screening followup program may contact you directly to make sure you're aware of the test results.
If the test shows some sickle hemoglobin, a second blood test is done to confirm the diagnosis. The second test should be done as soon as possible and within the first few months of life.
The primary care doctor may send you to a hematologist for a second blood test. A hematologist is a doctor who specializes in blood diseases and disorders. This doctor also can provide treatment for sickle cell disease if needed.
Doctors also can diagnose sickle cell disease before birth. This is done using a sample of amniotic fluid or tissue taken from the placenta. (Amniotic fluid is the fluid in the sac surrounding a growing embryo. The placenta is the organ that attaches the umbilical cord to the mother's womb.)
Testing before birth can be done as early as 10 weeks into the pregnancy. This testing looks for the sickle hemoglobin gene, rather than the abnormal hemoglobin that the gene makes.
How Is Sickle Cell Anemia Treated?
Sickle cell anemia has no widely available cure. However, treatments can help relieve symptoms and treat complications. The goals of treating sickle cell anemia are to relieve pain; prevent infections, organ damage, and strokes; and control complications (if they occur).
Blood and marrow stem cell transplants may offer a cure for a small number of people who have sickle cell anemia. Researchers continue to look for new treatments for the disease.
Infants who have been diagnosed with sickle cell anemia through newborn screening are treated with antibiotics to prevent infections. Their parents are educated about the disease and how to manage it. These initial treatment steps have greatly improved the outcome for children who have sickle cell anemia.
Specialists Involved
People who have sickle cell anemia need regular medical care. Some doctors and clinics specialize in treating people who have the disease. Hematologists specialize in treating adults and children who have blood diseases or disorders.
Treating Pain
Medicines and Fluids
Mild pain often is treated at home with over-the-counter pain medicines, heating pads, rest, and plenty of fluids. More severe pain may need to be treated in a day clinic, emergency room, or hospital.
The usual treatments for acute (rapid-onset) pain are fluids, medicines, and oxygen therapy (if the oxygen level is low). Fluids help prevent dehydration, a condition in which your body doesn't have enough fluids. Fluids are given either by mouth or through a vein. Your doctor may prescribe antibiotics if you have an infection.
Treatment for mild-to-moderate pain usually begins with acetaminophen (Tylenol®) or nonsteroidal anti-inflammatory drugs (NSAIDs), such as ibuprofen.
If pain continues or becomes severe, stronger medicines called opioids may be needed. Talk with your doctor about the possible benefits and risks of taking strong pain medicine, especially if the medicine will be used for a long period.
Hydroxyurea
Severe sickle cell anemia can be treated with a medicine called hydroxyurea. This medicine prompts your body to make fetal hemoglobin. Fetal hemoglobin, or hemoglobin F, is the type of hemoglobin that newborns have.
In people who have sickle cell anemia, fetal hemoglobin helps prevent red blood cells from sickling and improves anemia.
Given daily, hydroxyurea reduces how often painful sickle cell crises and acute chest syndrome occur. Many people taking hydroxyurea also need fewer blood transfusions and have fewer hospital visits.
Doctors are studying the long-term effects of hydroxyurea on people who have sickle cell anemia. Early studies in children suggest that the medicine may help improve growth and preserve organ function, but this has not been proven.
Hydroxyurea can reduce the number of white blood cells in your blood. (These cells help fight infections.) This can lead to an increased risk of infections.
People who take hydroxyurea must have careful medical followup, including blood tests. The dose of this medicine may need to be adjusted to reduce the risk of side effects.
A doctor who has knowledge about hydroxyurea can tell you about the risks and benefits of taking this medicine.
Preventing Complications
Blood transfusions are commonly used to treat worsening anemia and sickle cell complications. A sudden worsening of anemia due to an infection or enlarged spleen is a common reason for a blood transfusion.
Some, but not all, people who have sickle cell anemia need regular blood transfusions to prevent life-threatening problems, such as stroke, spleen problems, or acute chest syndrome.
Having routine blood transfusions can cause side effects. Examples include allergic reactions and a dangerous buildup of iron in the blood (which must be treated). In general, the blood supply is fairly safe from infections such as hepatitis and HIV.
For more information, go to the Health Topics Blood Transfusion article.
Infections
Infections can be a major complication of sickle cell anemia throughout life, but especially during childhood. Often, infections can be prevented or treated.
To prevent infections in babies and young children, treatments include:
- Daily doses of antibiotics. Treatment may begin as early as 2 months of age and continue until the child is at least 5 years old.
- All routine vaccinations (including a yearly flu shot), plus the pneumococcal vaccine.
If your child has sickle cell anemia and shows early signs of an infection, such as a fever, you should seek treatment right away.
Adults who have sickle cell anemia also should have flu shots every year and get vaccinated against pneumonia.
Eye Damage
Sickle cell anemia can damage the blood vessels in the eyes and the retinas. The retinas are the thin layers of tissue at the back of the eyes. Regular checkups with an eye doctor who specializes in diseases of the retina can help detect eye damage.
Strokes
Stroke prevention and treatment are now possible for children and adults who have sickle cell anemia. Starting at age 2, children who have sickle cell anemia can have routine ultrasound scans of their heads. This is called transcranial Doppler (TCD) ultrasound. These scans are used to check the speed of blood flow to the brain.
TCD scans allow doctors to find out which children are at high risk of stroke. Doctors can treat these children with routine blood transfusions to reduce the risk of stroke.
A doctor who has knowledge about blood transfusions and sickle cell disease can tell you about the benefits and risks of this treatment.
Treating Other Complications
Acute chest syndrome is a severe and life-threatening complication of sickle cell anemia. If acute (sudden) failure of the liver and kidneys also occurs, it's called acute multiple organ failure.
Treatment for these complications usually occurs in a hospital and may include oxygen therapy, blood transfusions, antibiotics, pain medicine, and balancing body fluids.
Leg ulcers (sores) due to sickle cell anemia can be very painful. Ulcers can be treated with cleansing solutions and medicated creams or ointments.
Skin grafts may be needed if the leg ulcers are ongoing. Bed rest and keeping the legs raised to reduce swelling are helpful. If you have a lot of pain from leg ulcers, your doctor may recommend a strong pain medicine.
Gallbladder surgery may be needed if the presence of gallstones leads to gallbladder disease.
Priapism (a painful erection in males) can be treated with fluids, medicines, or surgery.
Regular Health Care for Children
Children who have sickle cell anemia need routine health care (just like children who don't have the disease). They need to have their growth checked regularly. They also need to get the routine shots that all children get.
All children younger than 2 years old should see their doctors often. Children who have sickle cell anemia may need even more checkups. After age 2, children who have sickle cell anemia may not need to see their doctors as often, but they usually still need checkups at least every 6 months.
These visits are a time for parents to talk with their child's doctor and ask questions about the child's care. Talk with your child's doctor about eye checkups and whether your child needs an ultrasound scan of the brain.
Until age 5, daily penicillin is given to most children who have sickle cell anemia. Doctors also give many children a vitamin called folic acid (folate) to help boost red blood cell production.
Young children who have sickle cell anemia should have regular checkups with a hematologist (a blood specialist).
New Treatments
Research on blood and marrow stem cell transplants, gene therapy, and new medicines for sickle cell anemia is ongoing. The hope is that these studies will provide better treatments for the disease. Researchers also are looking for a way to predict the severity of the disease.
Blood and Marrow Stem Cell Transplant
A blood and marrow stem cell transplant can work well for treating sickle cell anemia. This treatment may even offer a cure for a small number of people.
The stem cells used for a transplant must come from a closely matched donor. This usually is a close family member who doesn't have sickle cell anemia. This limits the number of people who may have a donor.
The transplant process is risky and can lead to serious side effects or even death. However, new transplant approaches may improve treatment for people who have sickle cell anemia and involve less risk.
Blood and marrow stem cell transplants usually are used for young patients who have severe sickle cell anemia. However, the decision to give this treatment is made on a case-by-case basis.
Researchers continue to look for sources of bone marrow stem cells—for example, blood from babies' umbilical cords. They also continue to look for ways to reduce the risks of this procedure.
For more information, go to the Health Topics Blood and Marrow Stem Cell Transplant article.
Gene Therapy
Gene therapy is being studied as a possible treatment for sickle cell anemia. Researchers want to know whether a normal gene can be put into the bone marrow stem cells of a person who has sickle cell anemia. This would cause the body to make normal red blood cells.
Researchers also are studying whether they can "turn off" the sickle hemoglobin gene or "turn on" a gene that makes red blood cells behave normally.
New Medicines
Researchers are studying several medicines for sickle cell anemia. They include:
- Decitabine. Like hydroxyurea, this medicine prompts the body to make fetal hemoglobin. Fetal hemoglobin helps prevent red blood cells from sickling and improves anemia. Decitabine may be used instead of hydroxyurea or added to hydroxyurea.
- Adenosine A2a receptor agonists. These medicines may reduce pain-related complications in people who have sickle cell anemia.
- 5-HMF. This natural compound binds to red blood cells and increases their oxygen. This helps prevent the red blood cells from sickling.
How Can Sickle Cell Anemia Be Prevented?
Sickle cell anemia is an inherited disease. If a person is born with it, steps should be taken to reduce its complications. (For more information, go to "Living With Sickle Cell Anemia.")
People who are at high risk of having a child with sickle cell anemia and are planning to have children may want to consider genetic counseling. A counselor can explain the risk (likelihood) of having a child who has the disease. He or she also can help explain the choices that are available.
You can find information about genetic counseling from health departments, neighborhood health centers, medical centers, and clinics that care for people who have sickle cell anemia.
Living With Sickle Cell Anemia
With good health care, many people who have sickle cell anemia can live productive lives. They also can have reasonably good health much of the time and live longer today than in the past. Many people who have sickle cell anemia now live into their forties or fifties, or longer.
If you have sickle cell anemia, it's important to:
- Adopt or maintain a healthy lifestyle
- Take steps to prevent and control complications
- Learn ways to cope with pain
If you have a child or teen who has sickle cell anemia, you can take steps to learn about the disease and help your child manage it.
Adopt or Maintain a Healthy Lifestyle
To take care of your health, you should adopt or maintain healthy lifestyle habits.
Follow a healthy diet. A healthy diet includes a variety of fruits, vegetables, and whole grains. It also includes lean meats, poultry, fish, beans, and fat-free or low-fat milk or milk products. A healthy diet is low in saturated fat, trans fat, cholesterol, sodium (salt), and added sugar.
Your doctor may suggest that you take folic acid (a vitamin) every day to help your body make new red blood cells. You also should drink at least 8 glasses of water every day, especially in warm weather. This will help prevent dehydration, a condition in which your body doesn't have enough fluids.
Your body needs regular physical activity to stay healthy. However, you should avoid exercise that makes you very tired. Drink lots of fluids when you exercise. Talk with your doctor about how much and what kinds of physical activity are right for you.
You also should get enough sleep and rest. Tell your doctor if you think you may have a sleep problem, such as snoring or sleep apnea. Sleep apnea is a common disorder in which you have one or more pauses in breathing or shallow breaths while you sleep.
Ask your doctor whether you can drink alcohol and what amount is safe for you. If you smoke, quit. Talk with your doctor about programs and products that can help you quit smoking. Also, try to avoid secondhand smoke.
For more information about how to quit smoking, go to the Health Topics Smoking and Your Heart article and the National Heart, Lung, and Blood Institute's "Your Guide to a Healthy Heart." Although these resources focus on heart health, they include general information about how to quit smoking.
Take Steps To Prevent and Control Complications
Along with healthy lifestyle habits, you can take other steps to prevent and control painful sickle cell crises. Many factors can cause sickle cell crises. Knowing how to avoid or control these factors can help you manage your pain.
You may want to avoid decongestants, such as pseudoephedrine. These medicines can tighten blood vessels, making it harder for red blood cells to move smoothly through the vessels.
Avoid extremes of heat and cold. Wear warm clothes outside in cold weather and inside of air-conditioned rooms. Don't swim in cold water. Also, be cautious at high altitudes; you may need extra oxygen.
If possible, avoid jobs that require a lot of heavy physical labor, expose you to extremes of heat or cold, or involve long work hours.
Don't travel in airplanes in which the cabins aren't pressurized (that is, no extra oxygen is pumped into the cabin). If you must travel in such an airplane, talk with your doctor about how to protect yourself.
Ongoing Care
Get a flu shot and other vaccines to prevent infections. You also should see your dentist regularly to prevent infections and loss of teeth. Contact your doctor right away if you have any signs of an infection, such as a fever or trouble breathing.
For people who have sickle cell anemia, just like for everyone else, regular medical care and treatment for health issues are important. Your checkups may include extra tests for possible kidney, lung, and liver diseases. See a sickle cell anemia expert regularly. Also, see an eye doctor regularly to check for damage to your eyes.
Learn the signs and symptoms of a stroke. They include a lasting headache, weakness on one side of the body, limping, and sudden changes in speech, vision, or hearing. If you have any of these symptoms, call 9–1–1 right away.
Get treatment and control any other medical conditions you have, such as diabetes.
Talk with your doctor if you're pregnant or planning to become pregnant. Sickle cell anemia can worsen during pregnancy. You'll need special prenatal care.
Women who have sickle cell anemia also are at increased risk for an early birth or a low-birth-weight baby. However, with early prenatal care and frequent checkups, you can have a healthy pregnancy.
Emotional Issues and Support
Living with sickle cell anemia may cause fear, anxiety, depression, and stress. It's important to talk about how you feel with your health care team. Talking to a professional counselor also can help. If you're feeling very depressed, your doctor may recommend medicines or other treatments that can improve your quality of life.
Joining a patient support group may help you adjust to living with sickle cell anemia. You can see how other people who have the same symptoms have coped with them. Talk with your doctor about local support groups or check with an area medical center.
Support from family and friends also can help relieve stress and anxiety. Let your loved ones know how you feel and what they can do to help you.
Learn Ways To Cope With Pain
Pain is different for each person. Pain that one person can live with is too much for another person. Work with your doctor to find ways to manage your pain.
You may need both over-the-counter and prescription medicines. Your doctor may prescribe strong pain medicines. If so, talk with him or her about how to safely use these medicines.
Other ways to manage pain include using a heating pad, taking a hot bath, resting, or getting a massage. Physical therapy might help ease your pain by helping you relax and strengthening your muscles and joints.
Counseling or self-hypnosis also may help. You may find that activities that keep your mind off the pain, such as watching TV and talking on the phone, are helpful.
Caring for a Child Who Has Sickle Cell Anemia
If your child has sickle cell anemia, learn as much about the disease as possible. This will help you recognize early signs of problems, such as fever or chest pain, and seek early treatment.
Sickle cell centers and clinics can give you information and counseling to help you handle the stress of coping with your child's disease.
Ongoing Care
Your child will need to see the doctor often for blood tests. The doctor also will check your child for any possible damage to his or her lungs, kidneys, and liver.
Talk with the doctor about your child's treatment plan, how often he or she needs checkups, and the best ways to help keep your child as healthy as possible.
Preventing Infections
To prevent infections, make sure your child gets all of the vaccines that his or her doctor recommends. (For more information on vaccines, go to "How Is Sickle Cell Anemia Treated?")
Good hygiene also can help prevent infections. Make sure your child washes his or her hands often. This will help lower the chances of getting an infection.
Call the doctor right away if your child has any signs of infection, such as fever or trouble breathing. Keep a thermometer on hand and know how to use it. Call a doctor if your child has a temperature above 101 degrees Fahrenheit (about
Preventing a Stroke
Know the signs and symptoms of a stroke so you can take action. Signs and symptoms include a lasting headache, weakness on one side of the body, limping, and sudden changes in speech, vision, or hearing. Changes in behavior also may be a sign of a stroke.
Talk with the doctor about whether your child needs regular ultrasound scans of the head (transcranial Doppler (TCD) ultrasound). These scans can show whether your child is at high risk of a stroke.
Calling the Doctor
Ask your child's doctor about when you should call him or her right away. For example, he or she may want you to call right away if your child has any signs of a stroke or infection. You also may need to call if your child has:
- Swelling of the hands or feet.
- Swelling of the stomach. If the spleen gets larger than normal, you may see or feel swelling below the ribs on the left side of the body. Your child may complain that the area feels tender.
- Pale skin or nail beds or a yellowish color of the skin or whites of the eyes.
- Sudden fatigue (tiredness) with no interest in his or her surroundings.
- An erection of the penis that won't go away.
- Pain in the joints, stomach, chest, or muscles.
- A fever.
School-aged children can often, but not always, take part in physical education or sports. However, your child's doctor should approve any activity. Ask the doctor about safe activities for your child.
Caring for a Teen Who Has Sickle Cell Anemia
Teens who have sickle cell anemia must manage their condition, while also dealing with the stresses of the teen years. These teens also face some specific stresses related to sickle cell anemia, including:
- Body-image problems caused by delayed sexual maturity.
- Coping with pain and fear of addiction from using strong pain medicines.
- Living with uncertainty. (Sickle cell anemia is unpredictable and can cause pain and damage to the body at any time.)
Teen support groups and family and individual counseling are ways to support teens who have sickle cell anemia.
Clinical Trials
The National Heart, Lung, and Blood Institute (NHLBI) is strongly committed to supporting research aimed at preventing and treating heart, lung, and blood diseases and conditions and sleep disorders.
NHLBI-supported research has led to many advances in medical knowledge and care. For example, this research has uncovered some of the causes of blood diseases, as well as ways to prevent and treat these diseases.
Many questions remain about blood diseases and conditions, including sickle cell anemia. The NHLBI supports an extensive research program to learn more about sickle cell anemia and find better ways to treat the disease and prevent complications. For example, NHLBI-supported research on sickle cell anemia includes studies that explore:
- How genetics play a role in the development of sickle cell anemia
- The best ways to manage pain and treat other complications of sickle cell anemia
- How certain medicines and other therapies can help treat sickle cell anemia and improve quality of life for people who have the disease
- How to improve the safety and effectiveness of blood and marrow stem cell transplants for the treatment of sickle cell anemia
Much of this research depends on the willingness of volunteers to take part in clinical trials. Clinical trials test new ways to prevent, diagnose, or treat various diseases and conditions.
For example, new treatments for a disease or condition (such as medicines, medical devices, surgeries, or procedures) are tested in volunteers who have the illness. Testing shows whether a treatment is safe and effective in humans before it is made available for widespread use.
By taking part in a clinical trial, you can gain access to new treatments before they're widely available. You also will have the support of a team of health care providers, who will likely monitor your health closely. Even if you don't directly benefit from the results of a clinical trial, the information gathered can help others and add to scientific knowledge.
If you volunteer for a clinical trial, the research will be explained to you in detail. You'll learn about treatments and tests you may receive, and the benefits and risks they may pose. You'll also be given a chance to ask questions about the research. This process is called informed consent.
If you agree to take part in the trial, you'll be asked to sign an informed consent form. This form is not a contract. You have the right to withdraw from a study at any time, for any reason. Also, you have the right to learn about new risks or findings that emerge during the trial.
For more information about clinical trials related to sickle cell anemia, talk with your doctor. You also can visit the following Web sites to learn more about clinical research and to search for clinical trials:
- http://clinicalresearch.nih.gov
- www.clinicaltrials.gov
- www.nhlbi.nih.gov/studies/index.htm
- www.researchmatch.org
For more information about clinical trials for children, visit the NHLBI's Children and Clinical Studies Web page.
Links to Other Information About Sickle Cell Anemia
NHLBI Resources
- "A Century of Progress: Milestones in Sickle Cell Disease Research and Care"
- Featured Researcher: Dr. Mark Robbins, "Feasibility of Expert System To Promote African American Blood Donation for Sickle Cell Disease"
- Questions and Answers About Sickle Cell Trait
- Reports on Sickle Cell Disease Workshops and Meetings
- Sickle Cell Disease Guidelines (in Development)
- Sickle Cell Disease Research & Care
- Sickle Cell Disease Resources
- Story of Success: Reducing the Burden of Sickle Cell Disease
Sickle Cell News
- NHLBI Commemorates 100th Anniversary of First Published Report of Sickle Cell Disease
- Stroke Prevention Study in Children With Sickle Cell Anemia, Iron Overload Stopped Early (NHLBI News Release, June 4, 2010)
Non-NHLBI Resources
- "Hydroxyurea Treatment for Sickle Cell Disease" (Office of Medical Applications of Research)
- For People of African, Mediterranean, or Southeast Asian Heritage: Important Information about Diabetes Blood Tests (National Institute of Diabetes and Digestive and Kidney Diseases)
- Sickle Cell Anemia (MedlinePlus)
- Sickle Cell Disease (Centers for Disease Control and Prevention)
Clinical Trials
- Children and Clinical Studies
- Clinical Trials (Health Topics)
- Current Research (ClinicalTrials.gov)
- NHLBI Clinical Trials
- NIH Clinical Research Trials and You (National Institutes of Health)
- ResearchMatch (funded by the National Institutes of Health)