


About FDA
Medical Device Pre-Market Programs: An Overview of FDA Actions
 Executive Summary
Executive Summary
Nearly two years ago, the FDA’s Center for Devices and Radiological Health (CDRH) recognized that, given the growing complexities of medical product development, we needed to re-evaluate and modernize our regulatory review processes in order to assure that patients had timely access to safe and effective medical devices.
At that time, CDRH began to undertake a new systematic approach to device regulation - one that continued to focus on protecting public health by assuring that devices are safe and effective, but also focused on promoting public health by facilitating device innovation.
Innovation became one of our top four strategic priorities.
This new approach required that we move away from the traditional misperception that safety/effectiveness and innovation are incompatible. Rather than focus on more regulation or less regulation, we began to focus on smart regulation - how to effectively achieve both aspects of our mission as both a regulator and a facilitator. The FDA helps create a regulatory environment that allows innovation to thrive, by eliminating undue regulatory obstacles and assuring consumer confidence that medical technology in the U.S. is safe and effective.
Our goal has been to move away from the external perspective of a swinging pendulum and realign our activities on the premise that safety/effectiveness and innovation are complementary, mutually supporting aspects of our mission.
In August 2010, following extensive public input, we released two reports that identified problems with our pre-market programs. The reports also proposed potential actions for us to take to address the underlying root causes.
The number one problem we found was insufficient predictability in our pre-market programs, which can create inefficiencies, increased costs for industry and the FDA, and delays in bringing safe and effective products to market. It also can create challenges for small start-up companies to get investor and venture capitalist funding for new, early-stage technologies, which are critical to assuring that new technology reaches patients safely and effectively.
We identified several root causes of these problems. They include very high reviewer and manager turnover at CDRH, (almost double that of FDA’s drug and biologics centers); insufficient training for staff and industry; extremely high ratios of front-line supervisors to employees; insufficient oversight by managers; CDRH’s rapidly growing workload, caused by the increasing complexity of devices and the number of submissions we review; unnecessary and/or inconsistent data requirements imposed on device sponsors; insufficient guidance for industry; and poor-quality submissions from industry.
We identified proposed solutions to these problems, and we sought public comment on those solutions. In January 2011, we announced a Plan of Action that included 25 specific actions we would take this year to improve the predictability, consistency, and transparency of our pre-market review programs. In February 2011, we announced our Innovation Initiative that included several proposals to help maintain the position of the U.S. as the world’s leader in medical device innovation, including the creation of a new approach for important, new technologies called the Innovation Pathway.
Since that time, we have announced additional improvements to implement.
To best serve patients, the medical device industry and the FDA must have the flexibility to be innovative, to be entrepreneurial, and, ultimately successful. To succeed, several things must occur:
- CDRH must continue making critical improvements to our device program, in particular the actions described below;
- Industry and CDRH must work together to assure that the Center receives high-quality submissions, which contain the information we need to make well-informed and timely decisions; and
- CDRH must have additional, adequate and stable resources to get the job done right and quickly - this is the subject of user fee reauthorization negotiations.
We believe that if these three things are accomplished we will provide the kind of value that patients deserve and have come to expect from the FDA - timely access to safe and effective devices that address their health care needs. And the medical industry will have the kind of predictable, consistent, transparent, and efficient pathways to market that spur continued innovation.
While we work with industry toward a reauthorization of user fees to provide additional funding, we have continued to move forward to do our part. As described below and in an accompanying chart, we have already made significant progress on the actions we committed to take. Once these policies and processes are finalized and implemented, we expect to see a significant and positive impact.
The actions we are taking can be grouped into three main areas of emphasis. Specifically, our actions seek to:
- Create a culture change toward greater transparency, interaction, collaboration, and the appropriate balancing of benefits and risks;
- Assure predictable and consistent recommendations, decision making, and application of the least burdensome principle; and
- Implement efficient processes and use of resources.
Each of the actions described in this document has either already been implemented or will be implemented, at least in part, within the first half of 2012. We are moving forward as quickly as possible but are mindful that as a public agency, we must allow time for submission and careful review of public comment. This transparency allows us to benefit from the consideration of broader perspectives. However, the trade-off is that it takes longer to implement.
Conclusion
Through our assessments and extensive public input we identified several problems with our pre-market programs, their root causes, and solutions. We have now embarked on an effort to make the necessary improvements to assure that our pre-market programs are predictable, consistent, transparent, and efficient. Specifically, we are in the process of: (i) creating a culture change toward greater transparency, interaction, collaboration, and the appropriate balancing of benefits and risks; (ii) assuring predictable and consistent recommendations, decision making, and application of the least burdensome principle; and (iii) implementing efficient processes and use of resources.
We remain committed to improving our pre-market programs, assuring that patients have timely access to safe and effective devices, including cutting-edge technologies, and helping the U.S. device industry to remain strong and innovative.
Medical Device Pre-Market Programs: An Overview of FDA Actions
Introduction and Background
Nearly two years ago, the FDA’s Center for Devices and Radiological Health (CDRH) recognized that, given the growing complexities of medical product development, we needed to re-evaluate and modernize our regulatory review processes in order to assure that patients had timely access to safe and effective medical devices.
At that time, CDRH began to undertake a new systematic approach to device regulation - one that continued to focus on protecting public health by assuring that devices are safe and effective, but also focused on promoting public health by facilitating device innovation.
Innovation became one of our top four strategic priorities.
This new approach required that we move away from the traditional misperception that safety/effectiveness and innovation are incompatible. Rather than focus on more regulation or less regulation, we began to focus on smart regulation - how to effectively achieve both aspects of our mission as both a regulator and a facilitator. The FDA helps create a regulatory environment that allows innovation to thrive, by eliminating undue regulatory obstacles and assuring consumer confidence that medical technology in the U.S. is safe and effective.
As part of our process to improve our internal systems, we first reached out to our stakeholders to hear their concerns and listen to their recommendations about our pre-market programs. This is what we heard: industry felt that inadequate predictability, consistency and transparency were stifling innovation and driving jobs overseas; and consumer groups, third-party payers, and some health care professionals believed that one of our pre-market pathways - the 510(k) program - did not provide adequate protection for American patients by letting unsafe products on the market nor generate sufficient information for practitioners and patients to make well-informed treatment and diagnostic decisions. In turn, CDRH employees expressed concerns that the 510(k) program had not adapted to the increasing complexity of devices and that poor quality 510(k) submissions, poor quality clinical studies conducted in support of Pre-Market Approval (PMA) applications, and an ever-growing workload strained an already overburdened pre-market program.
Our goal has been to move away from the external perspective of a swinging pendulum and realign our activities on the premise that safety/effectiveness and innovation are complementary, mutually supporting aspects of our mission.
We began two assessments of our pre-market programs - one focused on the 510(k) program and one that looked at how we use science in regulatory decision making, touching on aspects of several of our pre-market review pathways, such as our clinical trials program - to identify the problems, their root causes and the appropriate solutions. In addition, we contracted with the Institute of Medicine (IOM) to conduct an independent evaluation of our 510(k) program.
In August 2010, following extensive public input, we released two reports that identified problems with our pre-market programs. The reports also proposed potential actions for us to take to address the underlying root causes.
The number one problem we found was insufficient predictability in our pre-market programs, which can create inefficiencies, increased costs for industry and the FDA, and delays in bringing safe and effective products to market. It also can create challenges for small start-up companies to get investor and venture capitalist funding for new, early-stage technologies, which are critical to assuring that new technology reaches patients safely and effectively.
Signs of problems with our pre-market programs first arose in 2002-2003 with a steady increase each year in the percent of 510(k)s that receive an Additional Information Letter1 and in the number of review cycles. As a result, starting in 2004-2005, total review times for PMAs and 510(k)s steadily increased, and the percent of 510(k)s that resulted in a determination of substantial equivalence began to decrease. (See Appendix)
We identified several root causes of these problems. They include very high reviewer and manager turnover at CDRH, (almost double that of FDA’s drug and biologics centers); insufficient training for staff and industry; extremely high ratios of front-line supervisors to employees; insufficient oversight by managers; CDRH’s rapidly growing workload, caused by the increasing complexity of devices and the number of submissions we review; unnecessary and/or inconsistent data requirements imposed on device sponsors; insufficient guidance for industry; and poor-quality submissions from industry.
Through our internal assessments, we identified proposed solutions to these problems, and we sought public comment on those solutions. In January 2011, we announced a Plan of Action that included 25 specific actions we would take this year to improve the predictability, consistency, and transparency of our pre-market review programs. In February 2011, we announced our Innovation Initiative that included several proposals to help maintain the position of the U.S. as the world’s leader in medical device innovation, including the creation of a new approach for important, new technologies called the Innovation Pathway.
Since that time, we have announced additional improvements to implement.To best serve patients, the medical device industry and the FDA must have the flexibility to be innovative, to be entrepreneurial, and, ultimately successful. To succeed, several things must occur:
- CDRH must continue making critical improvements to our device program, in particular the actions described below;
- Industry and CDRH must work together to assure that the Center receives high-quality submissions, which contain the information we need to make well-informed and timely decisions; and
- CDRH must have additional, adequate and stable resources to get the job done right and quickly - this is the subject of user fee reauthorization negotiations.
We believe that if these three things are accomplished we will provide the kind of value that patients deserve and have come to expect from the FDA - timely access to safe and effective devices that address their health care needs. And the medical industry will have the kind of predictable, consistent, transparent, and efficient pathways to market that spur continued innovation.
Simply providing more resources won’t solve the problems with our pre-market programs. However, more resources are one key component to our and industry’s success in bringing safe and effective devices to market quickly and efficiently. Insufficient funding is at the root of or a contributing factor to several of these problems.
While we work with industry toward a reauthorization of user fees to provide additional funding, we have continued to move forward to do our part. As described below and in an accompanying chart, we have already made significant progress on the actions we committed to take. Once these policies and processes are finalized and implemented, we expect to see a significant and positive impact.
Overview of Actions CDRH is Taking to Improve Our Medical Device Pre-Market Programs
The actions we are taking can be grouped into three main areas of emphasis. Specifically, our actions seek to:
- Create a culture change toward greater transparency, interaction, collaboration, and the appropriate balancing of benefits and risks;
- Assure predictable and consistent recommendations, decision making, and application of the least burdensome principle; and
- Implement efficient processes and use of resources.
Although some of the specific actions described below would help achieve more than one of these goals, we placed each action into one the above three categories for purposes of this overview.
A Note about Timing of Implementation
Each of the actions described in this document has either already been implemented or will be implemented, at least in part, within the first half of 2012. We are moving forward as quickly as possible but are mindful that as a public agency, we must allow time for submission and careful review of public comment. This transparency allows us to benefit from the consideration of broader perspectives. However, the trade-off is that it takes longer to implement.
We believe the actions we are taking now will have a visible, positive impact within the coming year by: providing greater predictability on data requirements through guidance, reducing unnecessary and/or inconsistent data requests through training and policy and process changes, implementing policies that lead to appropriately balanced benefit-risk determinations, using external experts more extensively, creating incentives to conduct clinical studies first in the U.S., speeding up Investigational Device Exemption (IDE)2 and implementing the Innovation Pathway. approval decisions,
For example, by the end of October 2011 we will have issued our draft guidance on early feasibility studies and will allows companies to take advantage of the draft policy voluntarily on a case-by-case basis; by the end of November 2011 we will have issued and implemented our SOP to assure that a request for additional information will not be sent unless approved by the appropriate level manager; by the end of January 2012 we will have issued and implemented our SOP to assure greater consistency in the review of pre-market documents (e.g., IDEs, (510(k)s, PMAs) when review staff change during the review; by the end of March 2012 we will have analyzed the results of our pilot for the Network of Experts, made any appropriate changes to our SOP and agreement templates, and expanded participation in the Network by at least two other health care professional or scientific organizations; by the end of March 2012 we will have issued and implemented the final guidance on benefit-risk determinations; by the end of March 2012 we will begin to implement the Innovation Pathway 2.0 and as well as begin to make applicable changes to our other pre-market programs.
In addition, as part of the Innovation Pathway 2.0 project (see discussion below), we will develop metrics for the key drivers of performance and important outcomes.
I. Create a culture change toward greater transparency, interaction, collaboration, and the appropriate balancing of benefits and risks
We will create a culture change through:
- Better engagement with industry;
- Greater use of external experts;
- Implementing flexible, risk-based policies that appropriately balance benefits and risks and apply a more patient-centric approach;
- Establishing new ways of doing business that add value; and
- Setting clear expectations for CDRH staff.
CDRH’s staff is comprised of physicians, nurses, engineers, biologists, chemists, physicists, epidemiologists, statisticians, and other scientific experts dedicated to protecting and promoting the public health. They believe passionately in CDRH’s mission and are conscious that our task is to protect as well as promote public health. They share industry’s desire to bring safe and effective devices to market quickly because they are not only doctors, nurses, and other health care professionals but they, their families, and their friends are also patients.
With science evolving at breakneck speed, practitioners focusing more on evidence-based medicine, and patients becoming more involved in their own care, all members of the medical device ecosystem must refocus. We must fully embrace the paradigm that assuring the safety and effectiveness of devices is everyone’s job and the responsibility resides as much with industry, practitioners and patients as with the Agency. If all of us don’t do our part and if we don’t collaborate, then public health suffers.
By engaging more collaboratively with industry, patients, and outside experts, better explaining our thinking and decision making, establishing the right balance between benefits and risks, setting the right expectations, and creating new internal processes and pathways that get safe and effective devices to market more quickly and efficiently, we will create a more open, interactive, and flexible culture at CDRH. Many of the innovative devices that come across our desks are developed by small, entrepreneurial companies that are flexible, dynamic, nimble and quick. To keep up with these companies, we must become more entrepreneurial ourselves. These changes to our culture will also allow us to continuously adapt so to handle new technologies as they are presented to us.
1. Better engagement with industry
Actions we are taking to engage better with industry include:
- Improve Interactive Review - Interactive review3, when used appropriately, can reduce the number of formal requests for additional information, the number of review cycles, and, as a result, pre-market review times. Our goal is to complete our review as often as possible during the first cycle. However, CDRH’s use of interactive review has been inconsistent. In addition, sponsors have had, at times, unreasonable expectations for interactions with CDRH staff, such as calling staff weekly for status updates. To make interactive review an effective tool, we are working with industry representatives through user fee reauthorization negotiations to establish performance goals for early and substantive interactions. When industry, the FDA, and Congress agree on a user fee reauthorization package that includes interactive review goals we would begin to implement the agreed upon goals for interactive review in 2012 before reauthorizing legislation is enacted. In the interim, we are working with our managers to identify best practices and have set up a tracking system to better track its use.
- Improve Pre-Submission Meetings - The key to a timely review of an IDE or pre-market application or submission is to identify the data requirements up front and help sponsors resolve any relevant issues that arise from data they generate prior to an official submission. Pre-submission meetings, (which include pre-IDE meetings), can help provide sponsors with the necessary predictability for efficient device assessment and review. However, at times, industry and CDRH have found such meetings of limited value. Industry has complained of delays in scheduling meetings and of CDRH not providing clear advice or changing that advice without good reason. CDRH has struggled with sponsors not submitting the level of information necessary to provide useful advice, and sponsors not following the advice given.
For example, according to a survey conducted by PwC, less than a quarter of device companies that meet with the FDA follow the advice received during those meetings. To improve the value of pre-submission meetings, CDRH will issue guidance in the coming weeks that clarifies the roles and responsibilities of the sponsor and of CDRH, including what a sponsor should provide in advance of a meeting. The guidance will also make clear that if CDRH receives the necessary materials in advance of the meeting, we will provide written advice that will not change unless there are subsequent modifications to the device or its use (such as a new intended use) or new data that raises new significant issues (such as a previously unknown serious safety risk), which would warrant a change in our advice. In addition, the guidance will outline expected timeframes for the submission of materials and meeting schedules that we would adhere to if available resources permit. Already requests for pre-submission meetings over the past 5 years have nearly doubled. Given our current resources, it has become increasingly more difficult to schedule timely meetings. Current user fees do not cover pre-submission meetings.
2. Greater use of external experts
As device complexity continues to accelerate, CDRH must have greater human bandwidth to understand new technologies, act as a facilitator to bring safe and effective devices to market, and conduct our work in a timely, efficient manner that adds value. However, it is unreasonable to expect CDRH to have all the necessary expertise and experience, particularly to review emerging technologies. CDRH currently has the ability to use outside experts, but it is generally limited to advisory panels, which are time-consuming and resource-intensive. We believe it is important that we have more immediate access to external experts to supplement our knowledge base and help us quickly address important scientific questions. Actions we are taking to achieve this goal include:
- Leverage External Scientific Expertise by Developing a Network of Experts - In October 2011, CDRH announced it would start a pilot program with select health care professional and scientific professional organizations to rapidly identify appropriate experts to help Center staff resolve important scientific questions. The pilot will run through the end of 2011, after which we will make any appropriate modifications and expand the program to include other organizations. At that time, we released draft standard operating procedures (SOPs) to govern our use of this network.
Use of this network can help CDRH reach well-informed decisions more quickly, and provide more timely reviews of device submissions. However, to be able to make the best use of external experts, CDRH needs sufficient internal expertise to engage in productive dialogues with these outside experts. For example, if CDRH seeks to learn from the experiences of a neurosurgeon with a new technology, the best outcome is likely to occur if CDRH has an experienced neurosurgeon who can speak with the outside expert rather than a physician in another field or a scientist, such as an engineer. The issue of adequate staffing is discussed below and is the subject of user fee reauthorization negotiations. - Broaden CDRH Staff Experience with New Devices through an Experiential Learning Program - A deeper understanding of new technologies can help CDRH staff better focus their review of device submissions, reduce the need to request additional information during a review, make better benefit-risk determinations, and foster a more interactive and collaborative relationship with industry and the health care professional community. We are developing an Experiential Learning Program that will be launched in 2012. The program will provide staff with real-world experience as they visit manufacturers, research and health care facilities, and academic institutions. The extent of participation will depend on available financial and human resources to support travel and cover the workload for staff who are engaged in training. At present, we are piloting such visits to manufacturer facilities on a case-by-case basis.
3. Implementing flexible, risk-based policies that appropriately balance benefits and risks and apply a more patient-centric approach
Benefit-risk determinations are central to many critical decisions CDRH makes every day. These decisions are science-based, but differences of opinion are more likely to arise in the absence of a clear framework for how to use the science to reach decisions. This can lead to less predictability and consistency in CDRH decision making or unnecessary appeals by sponsors who incorrectly believe a particular decision is wrong because the approach we took was not transparent.
Establishing a framework for making benefit-risk determinations and clarifying the application of such criteria can lead to more productive interactions and more predictable and consistent outcomes. It can also help assure an appropriately balanced perspective on benefits and risks that is sufficiently flexible to accommodate new and established technologies.
We cannot avoid all differences of opinion among CDRH and sponsors—science is often uncertain. However, we can reduce the likelihood of disagreements by following a transparent, well-understood framework. To achieve this goal, we are taking the following actions:
- Issue Guidance on Making Benefit-Risk Determinations in Medical Device Pre-Market Review - On August 15, 2011, CDRH issued draft guidance on making benefit-risk determinations as a part of medical device pre-market decisions. The document describes for the first time the criteria CDRH uses to make such determinations to assure that CDRH and industry approach these decisions consistently using a commonly understood framework. In addition, CDRH staff would be required to complete a template addressing each applicable criterion and including it in the administrative record for an IDE, PMA, or 510(k), if appropriate, thereby facilitating a dialogue both within the Center and between CDRH staff and sponsors on what matters most in making benefit-risk determinations. The criteria and template would also result in greater predictability and consistency in CDRH decision making. Of note, the criteria take a patient-centric approach by calling for the consideration of patients’ tolerance for risk in applicable cases to assure that our decisions appropriately take into account the needs and desires of the ultimate beneficiary of the FDA pre-market program - patients. We are currently working with a large coalition of patient advocacy groups organized by the National Organization for Rare Disorders on establishing mechanisms for obtaining reliable information on patient perspectives.
- Issue Guidance on Early Feasibility Studies - Providing appropriate incentives to conduct early feasibility studies first in the U.S. while not placing study subjects at inappropriate risk could result in new devices coming to the U.S. first and staying in the U.S. because our clinicians would gain early experience with these technologies, and, therefore, companies would continue their clinical studies here to leverage that experience. As a result, some important new devices would come on the market sooner in the U.S. than they have in the past. In the coming weeks, CDRH will issue draft guidance to clarifywhen an early feasibility study, including first-in-human trials, can be conducted earlier during device development than previously had occurred and allow for select iterative design changes without seeking additional approval by the Center with the goal of encouraging companies to conduct clinical studies of their new technologies in the U.S. first.
- Issue Guidance on IDE Decisions - When a clinical study is necessary to inform device development or product marketing, setting the bar for approval in the right place is critical for the timely conduct of relevant, high quality studies and, therefore, the timely approval of safe and effective devices. For example, approving a pivotal clinical study that would not support marketing approval places patients at unnecessary risk while wasting the sponsor’s and the FDA’s time and resources. On the other hand, failing to approve or unnecessarily delaying approval of a pivotal clinical study because issues that did not need to be resolved at the time of approval had not yet been satisfactorily addressed can delay patients access to new technologies and cause sponsors and the FDA to incur unnecessary costs. In the coming weeks, CDRH will issue guidance that clarifies the types of decisions FDA may reach in approving an IDE and gives a general explanation of the reasoning behind such decisions and what they mean for sponsors. This guidance should give sponsors a better idea of the possible outcomes of an IDE submission and what they need to do in order to reach a timely and satisfactory decision. This guidance will also help assure that CDRH properly balances benefits and risks in approving IDEs. As a result, we expect to approve high quality IDEs more quickly.
4. Establishing new ways of doing business that add value
Sometimes, as in the case of device pre-market assessment and review, old paradigms can make it more difficult for those involved to engage in new ways of thinking. Just as industry needs the flexibility to innovate, government, too, can be innovative through iterative experimentation. Actions we will take that improve the way we do business include:
- Create an Innovation Pathway - In February 2011 we proposed to establish a new pathway, and, in effect, a new approach to reviewing important devices - the Innovation Pathway. We started with an initial process, which we built as we went along. We piloted the process on a prosthetic arm controlled by the central nervous system. We chose this device because it was both a revolutionary technology with important public health implications for its currently intended use and future applications, and it would challenge CDRH’s current approach. It was an attempt to experiment and learn fast. Now we have embarked on developing the Innovation Pathway 2.0 with a far broader mandate. Rather than build onto the existing, flawed process or fix pieces here and there, we are looking at the entire system and developing a new pathway from the ground up based on past experience and new expectations. The focus is not on how to improve pre-market review but rather on how we can reduce the time and cost of device development, assessment, and review.. The effort is about taking a fresh look at how we assess risks in the context of probable benefits, how we engage early on with innovators, and how we create a program that is adaptable, sustainable, and value-adding. And, what we create for the Innovation Pathway will inform related improvements to our other pathways to market; in effect, it is an experimental, iterative process to challenge our programs at their foundations across all their applicable aspects. Therefore, the potential impact on the device program as a whole is significant. To achieve this goal and to do so quickly, we have assembled a team of entrepreneurs in residence - made up of external experts in medical device development, business process improvement, and information technology - who will work day-to-day with FDA staff and leadership to use innovative approaches that can rapidly build an improved version of the Innovation Pathway.
5. Setting clear expectations for CDRH staff
CDRH staff are very talented and highly committed to accomplishing the FDA’s mission and want to see patients have timely access to safe and effective devices. According to an organizational assessment conducted by an Office of Personnel and Management contractor in 2010, CDRH has one of the most mission-dedicated staff in all of the Federal government (over 90% of survey respondents understood CDRH’s mission and were willing to go above their job duties to achieve it). In the past the faults of our program have not been with our staff but rather with management for not providing them with the necessary direction and leadership.
We can best serve patients, industry, practitioners, and our staff by providing our staff with a clear view of what are management’s vision and priorities and assuring that management is focused on the actions that have the greatest impact on our core activities, such as pre-market review and post-market safety. Therefore, we will:
- Better Align Employee Performance Evaluations with CDRH’s Goals - If we are to create a culture change, we need to provide clear policies and processes for our staff and then align employee performance evaluations and incentives with the right activities or else we risk sending mixed messages to our staff. As part of our development of the Innovation Pathway 2.0 we are exploring those actions by CDRH that would have the greatest impact on assuring that devices are safe and effective and facilitating innovation, which could include appropriate data requests, timely and efficient reviews, and appropriately balanced benefit-risk determinations. Once identified we will incorporate those actions into the performance evaluations for CDRH staff with applicable responsibilities and target our incentives, such as promotion, towards successful accomplishment of those actions.
II. Assure predictable and consistent recommendations, decision making, and application of the least burdensome principle
In 1997, the Food and Drug Administration Modernization Act added two provisions that are know as “the least burdensome provisions” to the Federal Food, Drug, and Cosmetic Act. These statutory provisions refer to the evidence required: (1) to demonstrate the substantial equivalence of devices with differing technological characteristics, and; (2) to demonstrate effectiveness in a PMA application. In 2002, CDRH issued guidance further clarifying these provisions. CDRH remains committed to implementing the least burdensome statutory provisions and guidance.
We will assure predictability and consistency in our recommendations, decision making, and application of the least burdensome principle by:
- Providing adequate management oversight and staffing;
- Enhancing training;
- Improving internal processes;
- Adopting smarter policies and issuing more guidance; and
- Developing new communication tools.
We have learned through our own assessments and by reading industry-sponsored surveys that the greatest challenge sponsors face is insufficient predictability about what they must do to gain market approval (or clearance). We agree that greater predictability can: (i) reduce the time and resources it takes to assess and review technologies; (ii) make the process for sponsors and the FDA more efficient: (iii) get safe and effective medical devices to patients more quickly; and (iv) encourage venture capitalists and other sources of seed money to invest in early-stage U.S. device companies and products.
1. Providing adequate management oversight and staffing
- We cannot expect the most out of CDRH staff if we do not provide them with the support they need. Such support includes appropriate management, adequate training, and clear processes and policies. We also need a sufficient number and range of expertise of reviewers to efficiently handle our workload, which increased 27 percent from 2007 to 2010, and continues to grow.
At present, the ratio of front-line supervisors to staff is as high as 1:27. As a consequence, our staff members have too much work and managers cannot provide the appropriate level of oversight. Moreover, the turnover rate for CDRH staff is almost double that of FDA’s drug and biologics centers. We believe this is due to the higher workload and lower pay our reviewers and front-line managers receive as compared to comparable positions in other parts of the Agency and what they could make in the private sector. The result, which industry-sponsored surveys have repeatedly found and we have confirmed, is high staff turnover. And, high turnover leads to longer review times because when critical staff change during a review, particularly medical officers and front-line supervisors, it is a significant setback. High turnover rates also contribute to a workforce with limited pre-market review experience. Almost half of our review staff have 4 years or less experience as a reviewer and most front-line supervisors have 3 years or less experience as a manager.
Our drug and biologics centers encountered this same problem about a decade ago - high turnover rate, unsustainable workload, insufficient management support and high review times. Through appropriate increases in user fee funding and new retention incentive policies the turnover rate in the drug program dropped and has remained low.
Currently, this issue is the subject of medical device user fee reauthorization negotiations. Although the human drug user fee program is about 3 times the size of the device user fee program, with the drug center having roughly 5 times the number of physicians as CDRH and the drug program collecting about 10 times the amount in user fees as does the device program, we believe we could create a world class medical device program with far less.
2. Enhancing training
In the past, new CDRH staff learned how to review pre-market submissions through direct experience only. In September 2011, we launched a Reviewer Certification Program - a combination of required courses and auditing of work product - which all new reviewers must complete. The purpose of the program is to give reviewers the type of training that can help accelerate their learning curve and help them develop the skills and experience necessary to perform high quality reviews. If the program is successful, and if adequate resources are available, we will explore expanding the program or a modified version of it to include current review staff. Additionally, as new programmatic guidance documents are finalized, CDRH staff will be trained in the scope and application of the new guidances and training opportunities will be made available for industry.
3. Improving internal processes
High quality and consistent decision making requires that decisions are made with the appropriate input and at the appropriate level within CDRH. We are taking the following actions to assure we make the right decisions quickly and consistently:
- Establish a Center Science Council - On March 31, 2011, we established a Center Science Council - composed of CDRH senior leadership and experienced staff - to help assure consistency and predictability in our scientific decision making and to monitor the quality and performance of the device center’s scientific programs. The draft charter for the Council is available on our website. The Council addresses important scientific issues that warrant senior--level review prior to our taking action. For example, if a review team wants to increase the clinical data requirements for all manufacturers of a type of device, that proposed action is now brought to the Council to decide; whereas, in the past, the decision was made independently by the review team.
- Publish Standard Operating Procedures (SOP) on:
- When Additional Information can be Requested - The percentage of 510(k) submissions that receive an Additional Information (AI) Letter - the letters we send to companies when we have extensive questions in response to their submission - has been steadily increasing since 2002. In our analysis of 2010 AI Letters we determined that eight percent of requests in AI Letters were inappropriate. Making inappropriate requests for information to sponsors increases total review times and places an unnecessary burden on sponsors and on the FDA. By creating an SOP for when an AI Letter can be sent, the types of requests that can be made, and at what management level the decision must be made, we seek to reduce the number of inappropriate requests to as close to zero as possible.
- Change in Reviewer - As noted under number 2 above, CDRH has the highest level of reviewer turnover of all the medical product centers at the FDA - more than double that of the drug and biologics centers. We recognize that when a reviewer leaves, sponsors are negatively impacted. In an effort to minimize this adverse impact, we are developing an SOP to assure greater consistency in the review of pre-market documents (e.g., IDEs, PMAs, 510(k)s) when the lead reviewer changes during the course of a review or between the approval of an IDE and the review of a PMA or 510(k).
- Corrective and Preventive Actions (CAPA) for the Pre-Market Review Process - We have directed most of our actions towards improving our policies and procedures but no system can foresee all potential problems in advance. Therefore, we are implementing a CAPA system for pre-market review to identify, track, and correct or prevent problems. On October 1, 2011, we started a pilot of the new CAPA system in our Office of Device Evaluation.
4. Adopting smart policies and greater issuance of guidance
Issuing guidance documents and keeping them current can provide greater predictability by laying out for companies what steps to take in preparing a device application or submission for approval (or clearance). In the past, we generally issued 30 to 40 new or updated guidances annually, including special controls guidances for de novo approvals, which is not enough to keep up with the growing demand. As the need for new and updated guidances increases with the growth in types of devices, and as the rapidly changing scientific landscape makes current guidances outdated more quickly than in the past, the adverse impact of the Center’s limited capacity to issue guidances will only increase if we maintain the status quo.
We currently issue an annual list of guidances we propose to develop during the coming year and seek public comment on the list. In July 2011, we issued updated SOPs to further streamline our guidance development process. Last year we issued new SOPs on a pilot basis. Under the pilot we were able to increase our guidance document production by 22% in 2011 as compared to the year before. Also, we have implemented a tracking system to better manage guidance documents through the development and clearance process. In addition, we created a new position of Associate Director for Guidance and Policy who oversees the front-line management of the guidance development process. However, the staff who review applications are the same people who draft guidances, which limits their ability to devote significant time to develop guidance documents.
To significantly increase guidance document production further, CDRH would need a core team of technical writers, and managers, who could relieve review staff from their drafting responsibilities and we would need a sufficient number of review staff so they could spend time providing expert advice on guidance content to the technical writers while not adversely impacting pre-market application and submission reviews. Having the resources to expand our guidance development bandwidth is a subject of user fee reauthorization negotiations.
In the interim, CDRH is focused on issuing cross-cutting guidances that clarify critical aspects of our pre-market programs (including the guidances described above) and reinforce the appropriate application of the least burdensome principle. By the end of 2011, we will have issued guidance on:
- The 510(k) Process - Failure to adequately clarify key aspects of the 510(k) substantial equivalence standard has led to differences of opinion within CDRH and between CDRH and individual sponsors, resulting in inefficiencies due to unnecessary delays from attempts to resolve these differences or unnecessary data requests. This guidance will clarify critical issues, such as what constitutes a new intended use, when it is appropriate to use multiple predicates, and when we are likely to ask for clinical data.
- 510(k) Modifications - Science is constantly changing and new technologies continue to emerge. As a result, it may not be clear to sponsors when they do or do not need to submit a 510(k) for making a change to an already cleared device. In recent years there have been several notable cases wherein a sponsor should have submitted a 510(k) but did not due to lack of clarity in the submission criteria. This led to greater costs and uncertainty when they later learned of their mistake and avoidable loss of consumer confidence in their products. To provide greater predictability, on July 27, 2011, we issued an update to our current guidance on which modifications require and which do not require a new 510(k). This update focuses on newer technologies and trends, such as the use of software.
- Clinical Trials - One of the biggest drivers of longer review times for PMAs and non-approval decisions is poor quality clinical trials. Proper execution of adequately designed clinical studies is critical to a successful PMA. At the same time, requiring a sponsor to conduct a more robust, but more costly, clinical study that otherwise is not necessary to demonstrate that a particular device is safe and effective can delay the ultimate approval of that device or create sufficient disincentives that the technology is not brought to the U.S. market at all. To help address these problems, we issued draft guidance on August 15, 2011, to provide CDRH and sponsors with a common approach to designing clinical studies that incorporates the least burdensome principle so as to assure adequate (not highest) quality of clinical trials and expedite IDE and PMA decisions.
5. Developing new communication tools
We believe it is important to provide clear expectations for what data should be submitted to support a pre-market application or submission. Several of the actions described above, such as the new Center Science Council, will assure that data requirements are clearly established and only change when appropriate. And, if changes occur, they will be approved at the appropriate management level. However, it is equally important that we communicate a change in our expectations and the basis for that change to industry as soon as possible. In some cases, we need to implement a change right away to assure that a particular type of device is safe and effective. For example, if new data shows that a certain kind of implantable device can cause a serious harm we were not aware of before, such as strokes, it may be unethical, depending on the circumstances, to approve that type of device without first addressing the serious safety concern. Currently, device firms learn of such changes through new or updated guidance, which can take a year or two to develop. Or, even worse, they come in to talk with the Center when they submit an application, only to learn that they were using an outdated standard. This can lead to unnecessary costs and delays, and enormous frustration on the part of the sponsor. In response, CDRH has taken the following action:
- Create Notice to Industry Letters - On June 15, 2011, we issued an SOP for sending out Notice to Industry Letters. These letters are short communications that describe at a very high level changes to scientific data requirements and our reasons for those changes. Because these letters are short and are overseen by upper management at the Center, they can be developed and released more quickly than traditional guidances - roughly 3 weeks. The SOP limits the circumstances under which CDRH would change data requirements and immediately implement them. Also, each letter requires approval by the Center Science Council. Finally. although these letters are issued as final documents, we would provide an opportunity for public comment on these letters and we will change them in response to comments, if a change is warranted. We also recognize that providing meaningful comment on these letters could be challenging because of the limited discussion of data requirements. However, the alternative of first developing a fleshed out guidance document and seeking public comment before finalizing the document would either mean that we continue our current practice of notifying sponsors of a change once they submitted an application (or submission) to us, if we implemented the changed policy immediately, or we delay the review of those applications and submissions until we completed the guidance development process, which would add years to the review of some devices. Instead our proposed approach seeks to quickly implement and communicate a change in data requirements for a type of device, in the limited cases where fast action is warranted, while still providing an opportunity for public input.
III. Implement efficient processes and use of resources
Although additional and adequate resources are a critical component to the success of our pre-market programs, we must also make smart use of the resources we have and assure that our processes are optimally efficient. We are doing this by:
- Making existing processes more efficient;
- Using our resources more effectively; and
- Improving our ability to rely on data from outside the U.S. and actions by regulatory bodies of other countries.
1. Make Existing Processes More Efficient
Many of the actions we have announced create new processes or programs. However, we also have existing process that, if improved or streamlined, could significantly enhance pre-market review. We are taking the following actions to improve existing processes:
- Issue guidance on:
- The de novo Process - The de novo review program is supposed to provide a pathway to market for low-to-moderate-risk novel medical devices for which no predicate is available. Until now, the pathway has been underutilized because it is overly burdensome. For example, sponsors first must submit a 510(k) and receive a not substantially equivalent decision, as required by statute, before it can petition the FDA for a de novo determination. On September 30, 2011, we issued draft guidance to make the de novo process less bureaucratic, more timely, and more transparent and, thereby, a more viable pathway to market for novel low-to-moderate risk devices under the existing statutory framework. The guidance makes clear which devices are eligible for the de novo process, and what data are necessary to support de novo classification of suitable devices. If we are able to improve the de novo process, more low-to-moderate-risk novel devices will reach the market in a timely fashion. During 2011 we piloted some of the new approaches which sped up the de novo classification for 9 devices.
- External Appeals - Although issues between CDRH and sponsors optimally would be resolved informally, some matters may require resolution through a formal appeals mechanism. The Center’s current guidance has been criticized as not providing adequate clarity about options available to external parties to resolve differences of opinion with the Agency, and for being out of date regarding whom to contact. In the coming weeks, CDRH will issue updated guidance that clearly lays out the processes available for appealing a CDRH decision so that sponsors are aware of all of their options when they are not satisfied with particular review outcomes.
- Use Post-Market Data to Support Pre-Market Applications and Submissions - In some cases, robust post-market data could be used to reduce the need for pre-market data to support a pre-market application or submission, such as in the case of a modification to an already marketed device or the introduction of a new device that is similar to a device already on the market and made by the same manufacturer. CDRH has been actively engaged in facilitating the development of post-market surveillance systems. For example, we have participated in the establishment of over 25 registries, including the creation of an international consortium for orthopedic device registries. However, the U.S. lacks a national infrastructure for post-market data collection and analysis. The establishment of a unique device identification (UDI) system is a prerequisite before we can develop such an infrastructure because it would allow for the efficient linking of a specific device model with a patient’s experience with that device. Later this year we hope to issue a proposed regulation to establish a UDI system. However, much more would need to be done. The July 2011 Institute of Medicine’s report on the 510(k) program made several recommendations. One of them called for the development of a post-market surveillance strategy that would allow for the integration of post-market and pre-market data to help inform pre-market decisions. We plan to address this recommendation and the other recommendations in the report by the end of October 2011.
- Improve the Third Party Review Program - The Third Party Review Program was established to reduce the burden on FDA review staff by off-loading the review of lower-risk devices to third party contractors who had been trained to review 510(k)s for these lower-risk device types and, thereby, allow staff to focus more of their attention on higher-risk devices. However, the program has not reduced resource needs for CDRH because of limited use by industry and because poor quality reviews by some third parties necessitate that CDRH continue to closely scrutinize most submissions reviewed by third parties. According to an analysis conducted by CDRH in 2011, and consistent with a 2007 report to Congress, third party reviewers may err in their decisions due to insufficient training or expertise for the type of device reviewed or from lack of access to confidential information in the possession of CDRH that, if available, would have led to different actions or decisions by the third party reviewer. CDRH is currently exploring options to improve the program. Greater training and auditing would require additional resources but, combined with other improvements to the program, could increase both the quality of the reviews and the utilization of third party reviewers as well as reduce review times by eliminating the need for continuous CDRH oversight.
2. Use Our Resources More Effectively
Our existing resources are precious. We can do a better job of using them in a targeted way by focusing more attention on higher-risk products and less attention on lower-risk products, optimally leveraging national and international standards, and helping industry improve the quality of pre-market submissions. Specifically, our actions include:
- Down-Classification of Certain Well-Understood Devices - Some devices that are considered high-risk when they are first classified may be eligible for down-classification when, with greater experience and data, they are determined to pose only low-to-moderate risks. By down-classifying devices, when appropriate, CDRH can reduce unnecessary regulatory burdens on device makers without compromising patient safety. This also frees up resources for CDRH staff to focus on higher-risk devices. In July 2011, we de facto reclassified over thirty formerly high-risk devices to lower-risk classifications thus reducing the burden on our staff and industry and allowing these devices to reach patients more quickly and efficiently while still assuring their safety and effectiveness. We are also continuing to review currently marketed devices to determine if they should be down-classified. Unfortunately, formal down-classification requires, as a matter of statute, issuing a regulation, which is a resource-intensive undertaking for CDRH staff. This additional step makes it more difficult to reduce unnecessary regulatory burdens on industry.
- Implement a Process for Triaging Submissions - Not all submissions require the same level of scrutiny during review, even within the same review pathway. At present, submissions are assigned to reviewers on an availability basis as they come into the Center. In 2012, we will pilot a triage process in our Office of In Vitro Diagnostics first before expanding it. Devices will be triaged based on the anticipated extent of review and other relevant factors such as product risk and quality of submission. The goal of this process is to more efficiently use CDRH staff time and speed up the review process.
- Encourage the Appropriate Use of Consensus Standards - National and international consensus standards can give sponsors an excellent roadmap to use for certain aspects of device development and assessment thereby providing greater predictability while also reducing the resources necessary for CDRH review. By clarifying which consensus standards to use when, we hope to improve the efficiency of the review process and increase the likelihood of product approval or clearance for sponsors who comply with accepted consensus standards.
- Improve the Quality of Pre-Market Submissions - Earlier this year we conducted an analysis of 2010 AI Letters sent to sponsors to resolve unanswered questions or deficiencies in their 510(k) submissions. The analysis found that, while CDRH did on occasion inappropriately request additional information (see discussion above), most of the requests were for deficiencies the sponsors should have known how to avoid. For example, roughly 24 percent of the AI Letters were sent because the sponsor did not adopt the approach recommended in an FDA guidance nor did they use an alternative approach (as they are permitted to do). Reviewing poor quality submissions creates inefficiencies by having CDRH staff expend time and effort to address problems the sponsor should have been able to address prior to submission. Deficient submissions also increase review times because they create avoidable delays as these deficiencies get resolved. Unlike the FDA’s drug and biologics centers, CDRH does not return poor quality submissions to the sponsor. To address this problem and improve the efficiency of device review, CDRH is working with representatives of industry to establish reasonable objective criteria for when we would not accept a 510(k) submission.
- Implement an Assurance Case Pilot Program - Assurance cases have been used successfully by other industries, such as avionics, to efficiently minimize product risks and expedite government reviews. Under the assurance case approach a company identifies the risks its product would pose, the mitigations the company would implement to adequately minimize those risks, and discusses the evidence that supports the use of those mitigations. The logical structuring of an assurance case helps companies identify and address risks and makes it easier for government staff to effectively and efficiently review an application and avoid asking unnecessary questions. The assurance case gives the reviewer a roadmap through the 510(k) submission and allows the reviewer to see the big picture of how the sponsor has mitigated risks and reduced the likelihood of device error. On March 31, 2011, we started a pilot on the use of assurance cases for infusion pumps because of the widespread problems we had seen with these technologies over many years. Preliminary results suggest the use of an assurance case can reduce review times, at least for some infusion pump submissions. Depending on the results of this pilot it may be of benefit to both industry and CDRH to apply an assurance case approach, possibly only on a voluntary basis, to other types of devices if appropriate. We intend to make the results of the pilot available to the public and will seek public input first if we think there would be value to expanding the use of assurance cases.
In addition to the above actions, we are exploring the creation of standardized device-specific submission templates to help sponsors better organize their applications/submissions and make CDRH’s review more efficient because relevant information would be easier to find and digest.
3. Improve Our Ability to Rely on Data from Outside the U.S. and Actions by Regulatory Bodies of Other Countries
Medical device development occurs across the globe. Companies perform clinical trials all over the world. However, devices may be regulated by different countries in different ways. By aligning our U.S. regulations with those of other countries as appropriate, foreign data and actions, such as approval decisions and manufacturing facility inspections, may inform our pre-market reviews and expedite approval. Global harmonization or convergence is a long-term goal, but in the short-term, we are taking the following actions:
- Facilitate the Use of Clinical Studies Conducted Outside the U.S. - Device companies may first conduct clinical studies to support marketing approval in other countries. Although we would prefer that clinical studies be performed in the U.S., it may not always be practical even with the planned improvements to our IDE program. Ideally, companies will eventually conduct clinical studies that support global approval thereby reducing regulatory burdens in the U.S. and abroad. In 2012, we will issue a proposed rule that describes the circumstances under which we would rely on studies conducted in and for approval in other countries to minimize costs to industry and speed access to safe and effective devices for U.S. patients.
- Harmonize Regulatory Programs Among Nations - Harmonization (or convergence) of regulatory programs, including data requirements to support pre-market applications and submissions, as well as the sharing of information and best practices between countries can reduce regulatory burdens on industry, create efficiencies for CDRH, and increase patient access to important devices internationally. We strongly believe that there should be continued harmonization (and convergence) efforts and sharing of best practices between countries. For almost two decades the FDA has participated in the Global Harmonization Task Force (GHTF) along with the other founding member countries - the European Union, Canada, Australia, and Japan. Through GHTF we have produced many high-level policy documents. However, these documents have provided the greatest value to countries that are in the process of establishing a device regulatory system while providing limited value to the member countries because they are too high-level and do not address the complexities of implementation. Implementation of these policies and other harmonization activities will require regulators to share with their counterparts the operational aspects of their regulatory programs as well as privileged and confidential information. GHTF does not allow for these types of interactions between regulators. Moreover, due to the limited membership of GHTF, new harmonization efforts have been undertaken by other countries thereby increasing the likelihood of countries adopting disparate approaches to device regulation. To address these limitations, CDRH has been working with the other GHTF member countries to create a new forum for international medical device regulators with broader membership to focus on implementing harmonization (and convergence) activities while continuing to develop new and to update existing policy documents in collaboration with industry and other stakeholders. The first meeting to create the new forum took place in Washington, DC in February 2011. A second meeting took place in Ottawa, Canada in October 2011. We expect to hold the first meeting of the new forum in 2012 to which industry and other stakeholders will be invited to attend and will continue to play an active role.
Conclusion
Through our assessments and extensive public input we identified several problems with our pre-market programs, their root causes, and solutions. We have now embarked on an effort to make the necessary improvements to assure that our pre-market programs are predictable, consistent, transparent, and efficient. Specifically, we are in the process of: (i) creating a culture change toward greater transparency, interaction, collaboration, and the appropriate balancing of benefits and risks; (ii) assuring predictable and consistent recommendations, decision making, and application of the least burdensome principle; and (iii) implementing efficient processes and use of resources.
We remain committed to improving our pre-market programs, assuring that patients have timely access to safe and effective devices, including cutting-edge technologies, and helping the U.S. device industry to remain strong and innovative.
Appendix
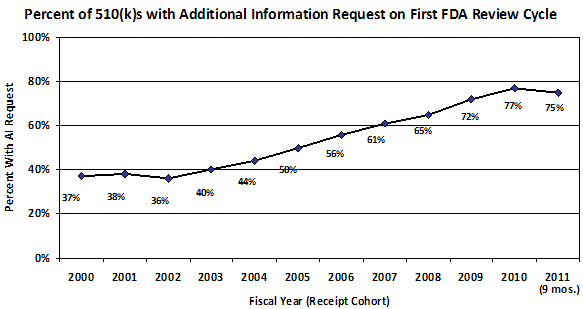
Figure 1. Percent of 510(k) submissions with Additional Information (AI) request on first FDA review cycle by fiscal year of receipt.
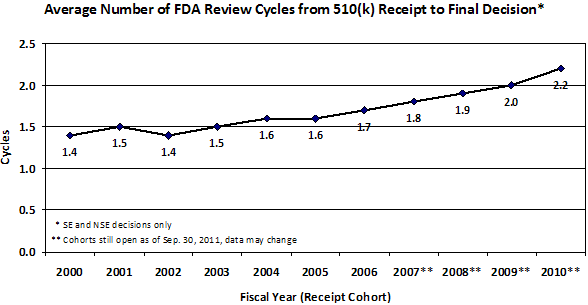
Figure 2. Average number of FDA review cycles from 510(k) receipt to final decision by fiscal year of receipt. As of September 30, 2011 some receipt cohorts are still open; data for those cohorts may change.
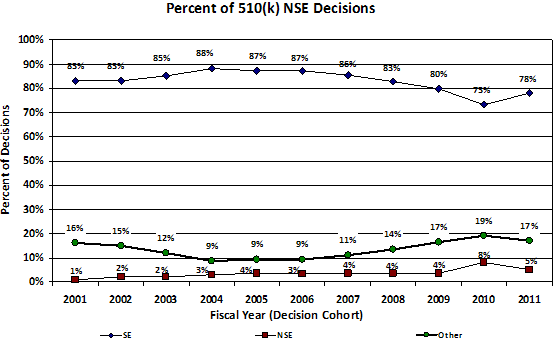
Figure 3. Percent distribution of 510(k) decisions by fiscal year of receipt.
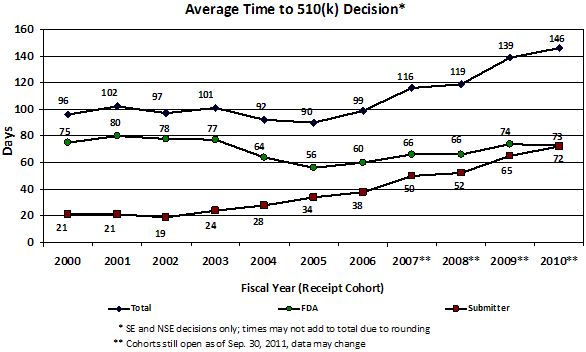
Figure 4. Average time to 510(k) decision by fiscal year of receipt.
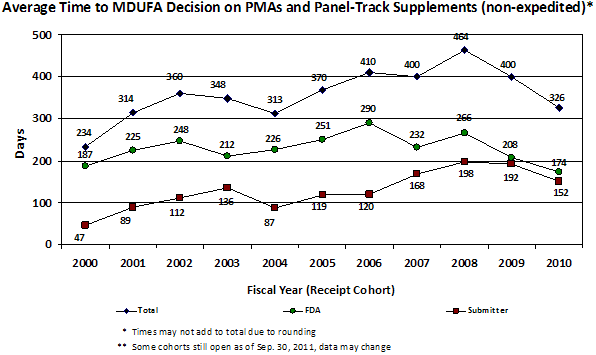
Figure 5. Average time to MDUFA decision for PMA and Panel-Track Supplements (non-expedited) by fiscal year of receipt. As of September 30, 2011 some receipt cohorts are still open; data for those cohorts may change.
1 When a submission contains insufficient information and a reviewer identifies a need for additional information, the reviewer will either call the submitter (Interactive Review) or prepare a letter outlining the additional information needed (Additional Information (AI) Letter). These letters include both formal letters sent via U.S. mail as well as “telephone hold” memos and e-mails. These letters include a comprehensive list of deficiencies associated with incoming original 510(k) submissions. Once an AI Letter is sent, the submission to which the letter pertains is placed on "hold" and is not considered to be under active review while the reviewer is waiting for a response. In other words, the clock stops during this time.
2 IDE refers to the regulations under 21 CFR 812. An approved IDE means that the IRB (and FDA for significant risk devices) has approved the sponsor’s study application and all the requirements under 21 CFR 812 are met. To conduct a clinical study of significant risk device the sponsor must first obtain FDA approval, which includes a review of the sponsor’s proposed study design, training documents, informed consent form, and other related materials. A significant risk device is an investigational device that: (1) is intended as an implant and presents a potential for serious risk to the health, safety, or welfare of a subject; (2) is for use in supporting or sustaining human life and represents a potential for serious risk to the health, safety, or welfare of a subject; (3) is for a use of substantial importance in diagnosing, curing, mitigating, or treating disease or otherwise preventing impairment of human health and presents a potential for serious risk to the health, safety, or welfare of a subject; or (4) otherwise presents a potential for serious risk to a subject.
3 See footnote 1.
