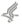

- Contact Us
 Get Email Alerts
Get Email Alerts
 Font Size
Font Size










| Home » Information for Researchers » Workshops, Meetings, and Scientific Reports » Lung |
Pulmonary Arterial HypertensionFuture DirectionsReport of a National Heart, Lung, and Blood Institute/Office of Rare Diseases WorkshopPublished in Circulation Volume 109. pp. 2947-2952, 2004 Internet address www.circulationaha.org John H. Newman, M.D., Barry L. Fanburg ,M.D., Stephen L. Archer, M.D., David B. Badesch, M.D., Robyn J. Barst, M.D., Joe G.N. Garcia, M.D., Peter N. Kao, M.D., Ph.D., James A. Knowles, M.D., Ph.D., James E. Loyd, M.D., Michael D. McGoon, M.D., Jane H. Morse, M.D., William C. Nichols, Ph.D., Marlene Rabinovitch, M.D., David M. Rodman, M.D., Troy Stevens, Ph.D., Rubin M. Tuder, M.D., Norbert F.Voelkel, M.D., and Dorothy B. Gail, Ph.D. From the Departments of Medicine, Nashville VA Medical Center (GRECC), and Vanderbilt University, Nashville, TN (JHN and JEL); Department of Medicine, New England Medical Center, Boston, MA (BLF); Division of Cardiology, University of Alberta Hospital, Edmonton, Alberta, Canada (SLA); Department of Medicine, University of Colorado Health Science Center, Denver, CO (DBB, DMR, and NFV); Pulmonary Hypertension Center, Columbia University College of Physicians & Surgeons New York, NY (RJB); Departments of Medicine and Pathology, Johns Hopkins University School of Medicine, Baltimore, MD (JGNG and RMT); Departments of Medicine/Pulmonary and Critical Care and Pediatrics, Stanford University Medical Center, Stanford, CA ( PNK and MR); Department of Medicine, Columbia University and College of Physicians and Surgeons, New York, NY (JAK and JM); Division of Cardiovascular Diseases, Mayo Clinic, Rochester, MN (MDM); Division and Program in Human Genetics, Cincinnati Children's Hospital Medical Center, Cincinnati, OH (WCN); Department of Pharmacology, Center for Lung Biology, University of South Alabama College of Medicine, Mobile, AL (TS); and the Division of Lung Diseases, NHLBI, Bethesda MD (DBG). This workshop, sponsored by the National Heart, Lung, and Blood Institute and the Office of Rare Diseases, National Institutes of Health, was held in Bethesda, Md., March 3-4, 2003 INTRODUCTION Pulmonary arterial hypertension (PAH) is characterized by vascular obstruction and the variable presence of vasoconstriction, leading to increased pulmonary vascular resistance and right heart failure. PAH can present in an idiopathic form, usually called primary pulmonary hypertension (PPH), and PAH is also associated with the scleroderma spectrum of diseases, HIV infection, portal hypertension with or without cirrhosis, and anorectic drug ingestion. Idiopathic PAH occurs in women more often than men (>2:1), has a mean age at diagnosis of 36 years, and is usually fatal within 3 years if untreated. Modern treatment has markedly improved physical function and has extended survival, and the five year mortality is about 50 percent. We still do not understand what initiates the disease or what allows it to progress. New studies of the pathogenetic basis of PAH will lead to targeted therapies for PAH. The National Heart, Lung and Blood Institute (NHLBI) and the Office of Rare Diseases (ORD), National Institutes of Health, convened a workshop to bring investigators together with various interests in vascular biology and pulmonary hypertension to identify new research directions. Discussion included genetics of PAH, receptor function, mediators, ion channels, extracellular matrix, signaling and potential clinical approaches. BACKGROUND AND QUESTIONS Molecular genetic studies have demonstrated mutations in a receptor in the transforming growth factor (TGF beta) superfamily, called bone morphogenetic protein receptor 2 (BMPR2), in most cases of familial pulmonary hypertension (1,2). Less common mutations associated with PAH occur in Alk1, a TGF receptor that also causes hereditary hemorrhagic telangectasia (3). Because only about 10-20% of persons with a BMPR2 mutation develop PAH, it is likely that other genes, genetic polymorphisms, and environmental factors are necessary to initiate the pathological sequence that leads to disease (4). Most cases of PAH are not associated with known inherited genetic mutations (5). Thus, external stimuli coupled with as yet undefined genetic susceptibility to disease are likely responsible for most cases of PAH. The abnormal transduction of signals related to BMPR2 and Alk/endoglin are unknown, and needs aggressive investigation. This will involve understanding of extracellular stimuli, receptor and membrane channel responses, and activation of a variety of intracellular molecules such as SMAD proteins, MAP kinases, and nuclear transcription factors (6). A number of basic questions are unanswered: 1. Is there a common final pathway that is activated in response to a variety of disease producing stimuli, or are there multiple independent pathways? 2. What cell or cells initiate the process in the vascular bed? 3. How does a mutated BMPR2 or Alk1 fail to suppress cellular abnormalities? 4. Why are females at higher risk for disease? and, 5. Are affected vessels subject to an autonomous process, or does the disease require ongoing stimuli, and therefore might be reversed? Multi-modality therapy needs to be considered for clinical study, especially with endothelin A receptor antagonists and phosphodiesterase 5 inhibitors. Drugs known to protect the systemic vascular bed such as the HMG Co-A reductase inhibitors are candidates for treatment trials. Other drugs, such as elastase inhibitors that have efficacy in experimental disease in animals need to be explored (7). PATHOGENESIS The accompanying Figure depicts the biological milieu from which abnormal pulmonary vascular responses might lead to primary pulmonary hypertension. The figure is necessarily simplified and serves only as a guide for discussion of pathogenesis. Discussion of potential therapies will interdigitate with mechanisms of disease.
Figure 1. This figure summarizes some of the cellular processes implicated in the pathogenesis of primary pulmonary hypertension. Extra-cellular mediators and cells (platelets) are highlighted in yellow, cell surface receptors and ion channels in purple, intra-cellular signaling in blue, and nuclear responses in green. See text for detailed descriptions of pathogenic mechanisms and the interactions among the many pathways that span the extra-cellular, membrane, cytosolic and nuclear domains. VEGF is vascular endothelial growth factor and its receptor is KDR. Intracellular transduction of this pathway is poorly understood. Endothelin is vasoactive and a mitogen, acting through Ca++ channels and ERK/Jun kinases. TIE is the angiopoietin receptor, a system found to be upregulated in pulmonary vascular disease (56). ALK1 and BMPR1-2 are receptors of the TGFb superfamily, and BMP is bone morphogenetic protein. ALK1 mutations cause hereditary hemorrhagic telangiectasia and some cases of PPH. EGF (epidermal growth factor), TNFa, ANGII (angiotensin II), and PDGF (platelet derived growth factor) are all proliferative stimuli that act through tyrosine kinase receptors and are partially transduced by intra-cellular oxidant species. In the intra-cellular domain, SMADs are regulatory proteins that activate nuclear transcription factors and interact with MAP kinases. AML1 is a nuclear transcription factor of potential importance. Elastase, downstream of AML1, has been implicated in vascular disease in experimental animals. Viral proteins are found in vascular lesions in the lungs of patients with PAH, raising the possibility that they participate in the pathogenesis (57). BMPR2 and Alk/endoglin mutations Multiple loss-of-function mutations have been described in BMPR2 and ALK1 (1,2,3,5). Activation of the TGF- BMPR2 axis leads to suppression of proliferation and activation of apoptosis; conversely, these loss-of-function mutations exaggerate the susceptibility of vascular cells to proliferate (8). Somatic mutations in endothelial cells microdissected from plexogenic lesions are found within the human MutS Homolog 2 gene that lead to reduced protein expression of TGF- and thus suppression of apoptosis (9). Furthermore, pulmonary artery smooth muscle cells (PASMC) from BMPR-knockout mice have abnormally enhanced proliferation rates in response to growth factors in vitro (10). The extensive diversity and tissue specificity of the SMAD system and the hetero-multimeric formation of different TGF/BMP receptor subtypes may explain the localization of the disease to the small pulmonary arteries. K+ channels, vascular tone and proliferation Vasoconstriction is a feature in some cases of pulmonary hypertension, and mechanisms relevant to PPH might exist in the pulmonary response to hypoxia. Hypoxia inhibits one or more voltage-gated potassium channels (Kv) in the pulmonary artery smooth muscle cells (PASMCs), opening voltage-gated calcium channels, raising cytosolic Ca2+ and initiating constriction (11). Kv1.5 or Kv2.1 channels are downregulated in the PASMC in humans with PAH (12) as well as in rats with chronic hypoxia-induced pulmonary hypertension (13). Furthermore, DNA microarray studies have shown downregulation of Kv channel genes in lungs of patients with PAH. In contrast, the genes for inward rectifier potassium channels (Kir) are upregulated in PAH (17). Whether these Kv channel abnormalities are genetically determined or acquired is unknown but they are not present in secondary pulmonary hypertension. It is unknown whether these PASMC Kv channel abnormalities are related to the TGFR2 abnormalities that have been described mostly in pulmonary artery endothelial cells (9). However, it is clear that the anorexigens, dexfenfluramine and aminorex, are K+ channel blockers (15). There is also a link between K+ channels and vascular remodeling through apoptosis, which may be relevant to PAH. Yuan et al have observed that agents that activate KCa and Kv channels, such as nitric oxide (NO), increase K+ efflux, which leads to cytosolic K+ loss, volume decrease and apoptosis (16). It has been hypothesized that PAH could also be viewed as a "K+ channelopathy" in which loss of channels (acquired or genetic) leads to vasoconstriction, cell proliferation and loss of basal apoptosis (17). Modulation of K+ channel function may have therapeutic potential. Augmenting the K+ channels should cause pulmonary vasodilatation and regression of pulmonary artery remodeling. Several oral treatments, such as dichloroacetate and sildenafil, may be able to enhance the function of these K+ channels (18). Sildenafil and other PD5 inhibitors cause pulmonary vasodilatation in large part through the BKCa mechanism. Oral dichloroacetate (DCA), a metabolic modulator, increases expression/function of Kv2.1 channels and decreases remodeling and PVR in rats with hypoxic pulmonary hypertension, partially via a tyrosine-kinase-dependent mechanism (18). DCA appears safe in humans (based on prior heart failure studies) and might be useful in treatment of PAH. Statins and mechanisms of PPH The HMG-CoA reductase inhibitors, statins, confer potent anti-proliferative and anti-inflammatory cardiovascular benefit in addition to cholesterol-lowering effects (19,20). Statins suppress endothelial and vascular smooth muscle cell neo-intimal responses to vascular injury in animal models (21,22). Among the mechanisms of statin actions is inhibition of the isoprenylation of rho and ras-family GTPases that couple membrane growth factor receptors to the intracellular MAP/ERK kinase signaling pathways that influence proliferation (23), Figure 1. Additionally, statins augment endothelium-dependent nitric oxide production and vasodilation through stabilization of endothelial nitric oxide synthase (eNOS) mRNA (24). Furthermore, statin enhancement of Akt kinase increases circulating endothelial progenitor cells that may contribute to vascular repair (25). In a monocrotaline rat model of pulmonary arterial hypertension, simvastatin attenuated and reversed both pulmonary hypertension and neointimal formation and improved survival from zero to 100%. Simvastatin reversed vascular occlusion through reduced intimal proliferation and increased apoptosis of pathological smooth muscle cells in pulmonary arteries (26). Similar results were recently noted in a rat model of hypoxic pulmonary hypertension (27). These data suggest that statins should be evaluated for treatment of patients with PPH and possibly for prevention in susceptible individuals. Elastase Inhibitors and Regression of Pulmonary Hypertension In rats subjected to hypoxia or monocrotaline, serine elastase increases in the pulmonary arteries prior to vascular remodeling, related to phosphorylation of MAP kinase and induction of AML1 transactivating activity (28,29). Inhibition of elastase attenuates the pulmonary hypertension and structural changes. Elastase activates matrix metalloproteinases, which amplify proteolytic response in the vessel wall and can release growth factors from the matrix in a biologically active form (30). The mitogenic potential of these growth factors is enhanced by elastase-MMP mediated induction of the glycoprotein, tenascin-C, via beta 3 integrin signaling (31). Tenascin amplifies the response to growth factors such as epidermal growth factor by inducing phosphorylation of growth factor receptors. In a monocrotaline rat model, elastase inhibition resulted in 86% survival versus 100% mortality, and regression of structural changes and pulmonary hypertension (32). Thus, elastase inhibitors may have promise in the treatment of clinical disease. Nitric Oxide NO is a potent pulmonary vasodilator, has anti-platelet activity, interacts with reactive oxygen species and protects K channel function (33). L-arginine is the sole substrate for nitric oxide synthase, and can be reduced by pregnancy or stress (34). Exogenous arginine seems to increase NO production. In endothelium, the arginine transporter is tightly co-localized with NO synthase (35). If the arginine transporter is disrupted by low level endothelial injury, extracellular levels of arginine might become insufficient. Arginine is an effective NO donor in the treatment of acute sickle cell lung crisis (36). Efforts to improve pulmonary hemodynamics in adults with pulmonary vascular disease with arginine have met with mixed results (37,38). Whether chronic arginine supplementation can improve the lung circulation in patients with PAH is unknown. It is possible that the co-administration of oral arginine or other NO donors with standard therapies would result in additive effects. Serotonin (5-hydroxytryptamine [5-HT]) 5-HT has been implicated in the pathogenesis of pulmonary arterial hypertension. Two most likely mechanisms are vasoconstriction and a mitogenic effect (39,40). Patients with PPH have decreased platelet 5-HT and increased plasma 5-HT concentration compared to controls, and increased release during platelet aggregation. Plasma 5-HT levels are also elevated in patients with fenfluramine induced pulmonary arterial hypertension (40). Dexfenfluramine releases 5-HT from platelets and inhibits reuptake, and causes inhibition of voltage-sensitive (Kv) channels, membrane depolarization and calcium entry into PASMC and megakaryocytes (15). These drugs are also serotonin transporter substrates and may interfere with intracellular signaling. The L-allelic variant of 5-HT transporter gene promoter, associated with 5-HT transporter over-expression and increased PASMC growth, is present in homozygous form in 65% of PPH patients and in 27% of controls. Thus, a 5-HT transporter polymorphism may confer susceptibility to PPH (40). 5-HT promotes PASMC hyperplasia through the serotonin transporter via production of reactive oxygen species and MAP kinase activation (41). PASMC from PPH patients grow faster than those from controls when stimulated by 5-HT due to increased expression of the serotonin transporter (40). In cultured rat PASMC, 5-HT potentiates the mitogenic effect of platelet-derived growth factor-BB. 5-HT transporter inhibitors eliminate the difference between PPH patients and controls in PASMC growth responses (41). Exploration of agents that inhibit 5-HT transporter (such as fluoxetine or paroxetine), reduce platelet aggregation and serotonin release (such as aspirin) or block serotonin receptors involved in vasoconstriction (such as ketanserin) in the management of PAH is warranted. Endothelin Endothelin, an endogenous peptide, is a powerful pulmonary vasoconstrictor and has mitogenic and fibrogenic effects (42). It is elevated in the blood in PAH. The vasoconstrictor effects are mediated by Ca++ channel activation and influx, and the growth and repair signals are transduced by activation of MAP kinases, including ERK and Jun via G-proteins (43). Endothelin receptor antagonists are highly effective in some patients with PAH and have become important therapies (42,43). The intracellular signaling pathways of endothelin interact with several other of the mediators of interest in PAH (Figure 1). Platelets and anti-platelet therapy Platelets present 5-HT, thromboxanes, and PDGF to the vascular wall, but there is very little known about the effects of anti-platelet therapy in PAH. Some of the long-term benefit of prostacyclin analogs (44-47) in the treatment of PAH might be from anti-platelet activity in addition to the vasodilator and possible inotropic effects. The clinical potential of primary anti-platelet agents has not been formally studied in PAH, in contrast to proven efficacy in systemic vascular disease. Aspirin therapy is particularly attractive in PAH, in part due to its reduction of platelet thromboxane production. Thromboxanes are elevated in patients with PAH (48), and prostacyclin and prostacyclin synthase are decreased in PAH (49). A favorable effect of anticoagulant therapy with warfarin in PAH is generally accepted, though based upon only three relatively small studies (50-52). It is unknown whether aspirin or other anti-platelet therapy would demonstrate similar benefit, with less risk and reduced need for monitoring. Genetic approaches PPH is a complex genetic disease, meaning that gene-gene and environment-gene interactions may confer susceptibility to disease. Approaches to discovering modifying genes will involve studies of PAH patients for underlying polymorphisms, such as in the serotonin transporter. A new approach is the development of a hypertensive phenotype in transgenic mice, either with hypoxia, drugs, other stimuli, or other underlying genetic backgrounds. The search for modifying genes will involve known candidate genes, such as NOS, but also genome wide surveys. Studies of the effects of polymorphisms are underway for many of the mediators shown in Figure 1. Genome wide searches using SNP analysis, RT PCR and microsatellite markers will require large cohorts and extensive resources to complete. Information from cDNA arrays and clusters and proteomic translation will be useful to determine the pathogenetic spectrum of disease in micro-dissected lesions and in stimulated cell and tissue experiments. At this time, little evidence about modifying genes has been published, so this field is young and full of promise. DISCUSSION REGARDING THERAPY Therapeutic advances over the past two decades have improved the natural history of PPH, and of PAH arising from other etiologies, including the scleroderma spectrum of disease, Eisenmenger's syndrome, HIV, anorectic drugs and portal hypertension. Current medical therapy includes: supportive treatment, e.g. digitalis, diuretics, supplemental oxygen; anticoagulation, (warfarin), calcium channel blockade (in the minority of patients with sustained vasodilation); chronic intravenous epoprostenol, newer PGI2 formulations, ( iv or inhaled iloprost, subcutaneous, aerosol and iv treprostinil, oral beraprost), (44-47), and endothelin receptor antagonists (42,43). Despite these advances, PAH remains a devastating disease and most approved therapies are very expensive and offer minor benefits to exercise capacity. Thus, there is a strong rationale to consider a number of novel therapies related to pathogenic mechanisms. These include, although are not limited to: phosphodiesterase inhibitors, statins, L-arginine, antiplatelet agents, serotonin inhibitors, agents to alter ion channel function, gene therapy, VIP (53), elastase inhibitors, anti-proliferative heparins (54), and possibly tyrosine kinase inhibitors. The use of multimodal/combination therapies may also further improve PPH treatment, but need critical evaluation in prospective, controlled trials. A major priority should be the discovery of new biomarkers that permit noninvasive diagnosis and monitoring of PPH and other forms of PAH. The workshop was not constituted to propose clinical trials, rather to find early leads for targeted therapy for future clinical applications. An excellent discussion of clinical trials in PPH has been recently published (55). FUTURE RESEARCH APPROACHES AND RECOMMENDATIONS The workshop discussions reflected the current momentum and excitement surrounding research on pulmonary arterial hypertension. The field is in a data gathering phase because of the application of new technology to the pulmonary circulation. New information will facilitate collaboration between basic scientists and clinical investigators and will accelerate translation to clinical care. Recommendations for research directions and opportunities are as follows: Genetic Studies
Receptors, Mediators, Ion Channels and Signaling Studies
Clinical Studies
REFERENCES
Dr Barst consults for Actelion Pharmaceuticals, Encysive Pharmaceuticals, Exhale Therapeutics, INO Therapeutics, Medtronic, Pfizer, and United Therapeutics Corp; serves on the advisory boards of Actelion Pharmaceuticals, Encysive Pharmaceuticals, Exhale Therapeutics, Pfizer, and United Therapeutics Corp; receives grant support from Actelion Pharmaceuticals, Encysive Pharmaceuticals, Exhale Therapeutics, INO Therapeutics, Medtronic, Myogen, Pfizer, and United Therapeutics Corp; and lectures for Actelion Pharmaceuticals and INO Therapeutics. Dr Badesch has received grant/research support from GlaxoWellcome, United Therapeutics, Boehringer Ingelheim, Actelion, ICOS/Texas Biotechnologies/Encysive, Pfizer, Myogen, American Lung Association, National Institutes of Health, and the Scleroderma Foundation. In addition, Dr Badesch has served as a consultant or speakers bureau member for GlaxoWellcome/GlaxoSmithKline, Actelion, Berlex, Astra-Merck, AstraZeneca, Myogen, Intermune, Forrest Labs, Encysive, and Exhale Therapeutics/CoTherix. Dr McGoon currently receives research funding from Myogen and Medtronic and has recently (in the past 5 years) received research funding from United Therapeutics, Pfizer, Actelion, GlaxoWellcome, and Boehringer Ingelheim. In addition, Dr McGoon has served on the data safety monitoring board of United Therapeutics Corp and has received honoraria from Myogen, Medtronic, United Therapeutics, Pfizer, and Actelion. Correspondence and requests for reprints should be addressed to: Dorothy B. Gail, Ph.D.
|
 Twitter
Twitter
 Facebook
Facebook YouTube
YouTube