
General Information About Adult Acute Lymphoblastic Leukemia (ALL)
Incidence and Mortality
Anatomy
Molecular Genetics
Diagnosis
Prognosis and Survival
Late Effects of Treatment for Adult ALL
ALL (also called acute lymphocytic leukemia) is an aggressive type of leukemia characterized by the presence of too many lymphoblasts or lymphocytes in the bone marrow and peripheral blood. It can spread to the lymph nodes, spleen, liver, central nervous system (CNS), and other organs. Without treatment, ALL usually progresses quickly.
Signs and symptoms of ALL may include the following:
- Weakness or fatigue.
- Fever or night sweats.
- Bruises or bleeds easily (i.e., bleeding gums, purplish patches in the skin, or petechiae [flat, pinpoint spots under the skin]).
- Shortness of breath.
- Unexpected weight loss or anorexia.
- Pain in the bones or joints.
- Swollen lymph nodes, particularly lymph nodes in the neck, armpit, or groin, which are usually painless.
- Swelling or discomfort in the abdomen.
- Frequent infections.
ALL occurs in both children and adults. It is the most common type of cancer in children, and treatment results in a good chance for a cure. For adults, the prognosis is not as optimistic. This summary discusses ALL in adults. (Refer to the PDQ summary on Childhood Acute Lymphoblastic Leukemia Treatment for more information about ALL in children.)
Incidence and MortalityEstimated new cases and deaths from ALL in the United States in 2012:[1]
- New cases: 6,050.
- Deaths: 1,440.
ALL presumably arises from malignant transformation of B- or T-cell progenitor cells.[2] It is more commonly seen in children, but can occur at any age. The disease is characterized by the accumulation of lymphoblasts in the marrow or in various extramedullary sites, frequently accompanied by suppression of normal hematopoiesis. B- and T-cell lymphoblastic leukemia cells express surface antigens that parallel their respective lineage developments. Precursor B-cell ALL cells typically express CD10, CD19, and CD34 on their surface along, with nuclear terminal deoxynucleotide transferase (TdT), while precursor T-cell ALL cells commonly express CD2, CD3, CD7, CD34, and TdT.
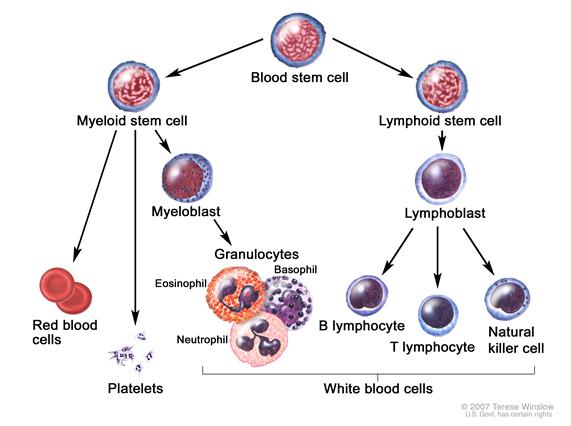
It has been recognized for many years that some patients presenting with acute leukemia may have a cytogenetic abnormality that is cytogenetically indistinguishable from the Philadelphia chromosome (Ph1).[3] The Ph1 occurs in only 1% to 2% of patients with acute myeloid leukemia (AML), but it occurs in about 20% of adults and a small percentage of children with ALL.[4] In the majority of children and in more than one-half of adults with Ph1-positive ALL, the molecular abnormality is different from that in Ph1-positive chronic myelogenous leukemia (CML).
Many patients who have molecular evidence of the bcr-abl fusion gene, which characterizes the Ph1, have no evidence of the abnormal chromosome by cytogenetics. The bcr-abl fusion gene may be detectable only by fluorescence in situ hybridization (FISH) or reverse-transcriptase polymerase chain reaction (RT-PCR) because many patients have a different fusion protein from the one found in CML (p190 vs. p210). These tests should be performed, whenever possible, in patients with ALL, especially in those with B-cell lineage disease.
L3 ALL is associated with a variety of translocations that involve translocation of the c-myc proto-oncogene to the immunoglobulin gene locus t(2;8), t(8;12), and t(8;22).
DiagnosisPatients with ALL may present with a variety of hematologic derangements ranging from pancytopenia to hyperleukocytosis. In addition to a history and physical, the initial workup should include:
- Complete blood count with differential.
- A chemistry panel (including uric acid, creatinine, blood urea nitrogen, potassium, phosphate, calcium, bilirubin, and hepatic transaminases).
- Fibrinogen and tests of coagulation as a screen for disseminated intravascular coagulation.
- A careful screen for evidence of active infection.
A bone marrow biopsy and aspirate are routinely performed even in T-cell ALL to determine the extent of marrow involvement. Malignant cells should be sent for conventional cytogenetic studies, as detection of the Ph1 t(9;22), myc gene rearrangements (in Burkitt leukemia), and MLL gene rearrangements add important prognostic information. Flow cytometry should be performed to characterize expression of lineage-defining antigens and allow determination of the specific ALL subtype. In addition, for B-cell disease, the malignant cells should be analyzed using RT-PCR and FISH for evidence of the bcr-abl fusion gene. This last point is of utmost importance, as timely diagnosis of Ph1 ALL will significantly change the therapeutic approach.
Diagnostic confusion with AML, hairy cell leukemia, and malignant lymphoma is not uncommon. Proper diagnosis is crucial because of the difference in prognosis and treatment of ALL and AML. Immunophenotypic analysis is essential because leukemias that do not express myeloperoxidase include M0 AML, M7 AML, and ALL.
The examination of bone marrow aspirates and/or biopsy specimens should be done by an experienced oncologist, hematologist, hematopathologist, or general pathologist who is capable of interpreting conventional and specially stained specimens.
Prognosis and SurvivalFactors associated with prognosis in patients with ALL include the following:
- Age: Age, which is a significant factor in childhood ALL and AML, may be an important prognostic factor in adult ALL. In one study, overall, the prognosis was better in patients younger than 25 years; another study found a better prognosis in patients younger than 35 years. These findings may, in part, be related to the increased incidence of the Ph1 in older ALL patients, a subgroup associated with poor prognosis.[5,6]
- CNS involvement: As in childhood ALL, adult patients with ALL are at risk of developing CNS involvement during the course of their disease. This is particularly true for patients with L3 (Burkitt) morphology.[7] Both treatment and prognosis are influenced by this complication.
- Cellular morphology: Patients with L3 morphology showed improved outcomes, as evidenced in a completed Cancer and Leukemia Group B study (CLB-9251 [NCT00002494]), when treated according to specific treatment algorithms.[8,9] This study found that L3 leukemia can be cured with aggressive, rapidly cycling lymphoma-like chemotherapy regimens.[8,10,11]
- Chromosomal abnormalities: Chromosomal abnormalities, including aneuploidy and translocations, have been described and may correlate with prognosis.[12] In particular, patients with Ph1-positive t(9;22) ALL have a poor prognosis and represent more than 30% of adult cases. Bcr-abl-rearranged leukemias that do not demonstrate the classical Ph1 carry a poor prognosis that is similar to those that are Ph1-positive. Patients with Ph1-positive ALL are rarely cured with chemotherapy, although long-term survival is now being routinely reported when such patients are treated with combinations of chemotherapy and Bcr-abl tyrosine kinase inhibitors.
Two other chromosomal abnormalities with poor prognosis are t(4;11), which is characterized by rearrangements of the MLL gene and may be rearranged despite normal cytogenetics, and t(9;22). In addition to t(4;11) and t(9;22), compared with patients with a normal karyotype, patients with deletion of chromosome 7 or trisomy 8 have been reported to have a lower probability of survival at 5 years.[13] In a multivariate analysis, karyotype was the most important predictor of disease-free survival.[13][Level of evidence: 3iiDii]
Long-term follow-up of 30 patients with ALL in remission for at least 10 years has demonstrated ten cases of secondary malignancies. Of 31 long-term female survivors of ALL or AML younger than 40 years, 26 resumed normal menstruation following completion of therapy. Among 36 live offspring of survivors, two congenital problems occurred.[14]
References
- American Cancer Society.: Cancer Facts and Figures 2012. Atlanta, Ga: American Cancer Society, 2012. Available online. Last accessed September 24, 2012.
- Pui CH, Jeha S: New therapeutic strategies for the treatment of acute lymphoblastic leukaemia. Nat Rev Drug Discov 6 (2): 149-65, 2007. [PUBMED Abstract]
- Peterson LC, Bloomfield CD, Brunning RD: Blast crisis as an initial or terminal manifestation of chronic myeloid leukemia: a study of 28 patients. Am J Med 60(2): 209-220, 1976.
- Secker-Walker LM, Cooke HM, Browett PJ, et al.: Variable Philadelphia breakpoints and potential lineage restriction of bcr rearrangement in acute lymphoblastic leukemia. Blood 72 (2): 784-91, 1988. [PUBMED Abstract]
- Gaynor J, Chapman D, Little C, et al.: A cause-specific hazard rate analysis of prognostic factors among 199 adults with acute lymphoblastic leukemia: the Memorial Hospital experience since 1969. J Clin Oncol 6 (6): 1014-30, 1988. [PUBMED Abstract]
- Hoelzer D, Thiel E, Löffler H, et al.: Prognostic factors in a multicenter study for treatment of acute lymphoblastic leukemia in adults. Blood 71 (1): 123-31, 1988. [PUBMED Abstract]
- Kantarjian HM, Walters RS, Smith TL, et al.: Identification of risk groups for development of central nervous system leukemia in adults with acute lymphocytic leukemia. Blood 72 (5): 1784-9, 1988. [PUBMED Abstract]
- Lee EJ, Petroni GR, Schiffer CA, et al.: Brief-duration high-intensity chemotherapy for patients with small noncleaved-cell lymphoma or FAB L3 acute lymphocytic leukemia: results of cancer and leukemia group B study 9251. J Clin Oncol 19 (20): 4014-22, 2001. [PUBMED Abstract]
- Hoelzer D, Ludwig WD, Thiel E, et al.: Improved outcome in adult B-cell acute lymphoblastic leukemia. Blood 87 (2): 495-508, 1996. [PUBMED Abstract]
- Fenaux P, Lai JL, Miaux O, et al.: Burkitt cell acute leukaemia (L3 ALL) in adults: a report of 18 cases. Br J Haematol 71 (3): 371-6, 1989. [PUBMED Abstract]
- Reiter A, Schrappe M, Ludwig WD, et al.: Favorable outcome of B-cell acute lymphoblastic leukemia in childhood: a report of three consecutive studies of the BFM group. Blood 80 (10): 2471-8, 1992. [PUBMED Abstract]
- Chromosomal abnormalities and their clinical significance in acute lymphoblastic leukemia. Third International Workshop on Chromosomes in Leukemia. Cancer Res 43 (2): 868-73, 1983. [PUBMED Abstract]
- Wetzler M, Dodge RK, Mrózek K, et al.: Prospective karyotype analysis in adult acute lymphoblastic leukemia: the cancer and leukemia Group B experience. Blood 93 (11): 3983-93, 1999. [PUBMED Abstract]
- Micallef IN, Rohatiner AZ, Carter M, et al.: Long-term outcome of patients surviving for more than ten years following treatment for acute leukaemia. Br J Haematol 113 (2): 443-5, 2001. [PUBMED Abstract]
 Back to Top
Back to Top