Each of the interactions commonly employed in the potential energy function ![]() is sketched below. Simple harmonic terms
describe bond stretching and angle bending. The planarity of groups (e.g., the amide planes of proteins) can also
be enforced by harmonic potentials known as an improper dihedrals. Rotation about single bonds (torsions)
is governed by sinusoidal energies.
is sketched below. Simple harmonic terms
describe bond stretching and angle bending. The planarity of groups (e.g., the amide planes of proteins) can also
be enforced by harmonic potentials known as an improper dihedrals. Rotation about single bonds (torsions)
is governed by sinusoidal energies.
The electrostatic attraction or repulsion between two charges is described by Coulomb's law:

A distance-dependent dielectric coefficient (RDIE:
![]() ) has been used to approximate solvent screening
without including explicit water molecules. Physically, it's a pretty ugly way to cheat. But if you don't want to include water,
it may be the best your simulation package has to offer; it is almost certainly better than using unscreened partial charges in the absence
of water. For realistic dynamics, we recommend constant-dielectric (CDIE) simulations with explicit solvation and
) has been used to approximate solvent screening
without including explicit water molecules. Physically, it's a pretty ugly way to cheat. But if you don't want to include water,
it may be the best your simulation package has to offer; it is almost certainly better than using unscreened partial charges in the absence
of water. For realistic dynamics, we recommend constant-dielectric (CDIE) simulations with explicit solvation and
![]() . The
presence of water retards conformational searching, however.
. The
presence of water retards conformational searching, however.
An important electrodynamic effect remains to be included: van der Waals interactions.
The electron cloud of a neutral atom fluctuates about the positively charged nucleus.
The fluctuations in neighboring atoms become correlated, inducing attractive dipole-dipole interactions.
The equilibrium distance between two proximal atomic centers is determined by a trade off between
this attractive dispersion force and a core-repulsion force that reflects electrostatic repulsion and the Pauli exclusion principle.
The Lennard-Jones potential models the attractive interaction as
![]() and the repulsive one as
and the repulsive one as
![]() :
:
![\begin{displaymath}
V_{ij}^{LJ}(r_{ij}) = \varepsilon_{ij} \left[ \left( \frac{r...
...{12} - 2 \left( \frac{r_{ij}^{min}}{r_{ij}} \right)^6 \right],
\end{displaymath}](img137.gif)
Perhaps, you are thinking, `Hey, what about magnetic forces?' The magnetic force between two moving charges is expressed
in terms of a double vector cross product involving the two particle velocities and the vector ![]() of separation. It does
not generally act along
of separation. It does
not generally act along ![]() , but it does when two charges q have instantaneous velocities v along parallel lines.
For this case, we can conveniently compare the magnitudes of the magnetic and electric forces.
It turns out that the magnetic force is weaker than the electric force by a factor of
, but it does when two charges q have instantaneous velocities v along parallel lines.
For this case, we can conveniently compare the magnitudes of the magnetic and electric forces.
It turns out that the magnetic force is weaker than the electric force by a factor of ![]() , where c is the speed of light.
Thus, magnetic forces are neglible for nonrelativistic particles,
such as the partial charges that are used in simulation force fields. For example, if a particle moves as much as 1 Å in as short
a time as 1 femtosecond (
, where c is the speed of light.
Thus, magnetic forces are neglible for nonrelativistic particles,
such as the partial charges that are used in simulation force fields. For example, if a particle moves as much as 1 Å in as short
a time as 1 femtosecond (![]() s), then
s), then
![]() . We may therefore completely neglect magnetic interactions.
. We may therefore completely neglect magnetic interactions.
 |
|---|
| Interactions included in representative potential energy function for MD simulation. |
For those who like equations with their pictures, a typical potential energy function used in MD simulations looks like:
![\begin{displaymath}
V_{bonded}(\vec{R}) = \sum_{bonds} k_l (l - l_0)^2 + \sum_{a...
...^2+ \sum_{torsions} A_n \left[ 1 + cos(n\phi - \phi_0) \right]
\end{displaymath}](img152.gif)
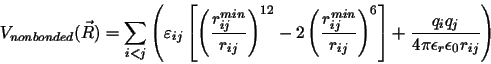
The first `bonded' sum is over bonds between atom pairs; the second sum is over bond angles defined by three atoms; the third and fourth sums are over
atom foursomes (as in the figure above). For bookkeeping purposes, each atom is assigned a number.
In the `nonbonded' interactions (van der Waals and electrostatics), the summation is over atoms i and j,
where `i < j' simply ensures that each interaction is counted only once.
Generally, atoms separated by one or two bonds are excluded
from the nonbonded sum, and those separated by three bonds, `1-4 interactions', may have electrostatic interactions reduced
by a multiplicative scale factor.
The form of ![]() shown here reflects the choice not to include an explicit hydrogen bond term, favoring instead to account for hydrogen bonds
through an appropriate parameterization of Lennard-Jones and Coulomb interactions. Note also that a single dihedral angle (torsion) may
have an energy described by more than one Fourier component (multiple values of n).
shown here reflects the choice not to include an explicit hydrogen bond term, favoring instead to account for hydrogen bonds
through an appropriate parameterization of Lennard-Jones and Coulomb interactions. Note also that a single dihedral angle (torsion) may
have an energy described by more than one Fourier component (multiple values of n).