
Alejandro A. Schäffer
NCBI
e-mail: schaffer@helix.nih.gov
Customer service through CIT/Division of Computational Biology (CIT/DCB):
Jim Tomlin: jtomlin@helix.nih.gov
http://bimas.cit.nih.gov/linkage/index.html
Download a copy of these course overheads in PostScript format
Caution: All conclusions are obtained with high probability, but not certainty
Reduce easy cases of linkage analysis to:
TO: J. D. Terwilliger and J. Ott, Handbook of Human Genetic Linkage, Johns Hopkins Press, 1994.
JO: Analysis of Human Genetic Linkage, Johns Hopkins Press, 1991
LD: LINKAGE Documentation
FD: FASTLINK Documentation
AD: APM Documentation
CD: GenoCheck Documentation
GD: GENEHUNTER Documentation
SD: SimIDB Documentation
VD: VITESSE Documentation
Parametric:
Non-parametric:
[S.A.G.E. sages: Joan Bailey-Wilson and Alexander Wilson, at NHGRI.]
Parametric means specify model of inheritance
many disease studies still use LINKAGE
Reason: TO and associated mini-courses
Suppose loci A and B are conjectured to be on the same chromosome.
Individual i is a recombinant w.r.t. parent p, if i inherits the A allele from one chromatid of p and the B allele from the other chromatid of p.
Recombination Fraction:
Probability that a child is recombinant.
between loci A and B is
 .
.
Low prob. recombinant ![]() linkage
linkage
define a vector 
which is an ordered list of recombination fractions between adjacent loci
Recombination fractions vary for male/female parents, but published maps tend to be gender-averaged
Genetic Distance  is the expected number
of crossovers between locus A and locus B.
is the expected number
of crossovers between locus A and locus B.
If A and B are on different chromosomes,
 , but
is not defined.
, but
is not defined.
 , while
may be larger than 1.
, while
may be larger than 1.
On the same chromosome,  and they are close for small values.
and they are close for small values.
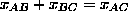

x is measured in Morgans, is unitless
1 Morgan means 1 expected crossover
Relationship between Morgans and Megabases is poorly understood.
Mapping functions convert from to x
and back.
Canonical application: compute
 knowing
and
knowing
and

and 

Published maps show between adjacent markers,
LINKAGE needs , but you may not
want to use loci adjacent on map.
JO: Chapter 1 and FD: README.mapfun
Given a fixed order of markers and disease loci, e.g.:
 D
D 

and a candidate vector,
compute a number called the ``likelihood'' to quantify:
How well does the data fit the theta?
Higher likelihood is better.
New versions of LINKAGE main programs
Program to replace MLINK and LINKMAP
``Simple'' means each person has <=2 grandparents in pedigree, and no loops.
J. Tomlin has a utility program to combine FASTLINK (LINKMAP) and VITESSE output for mixed-simple data sets.
Reference: VD
Compare two candidates:  ,
, 
 : is the correct value
: is the correct value
 : is the correct value
: is the correct value
Compute :
)/Like()
If this ratio is above a prescribed threshold, then
accept , reject ; if below a different
threshold, accept , reject .
Thresholds depend on the bad consequences to society from false negatives and false positives.
Unlinked means that  between the disease and
all other markers is 0.5
between the disease and
all other markers is 0.5
Linked means that between the disease and
some other marker is (substantially)  .
Use the best choice for hypothesis .
.
Use the best choice for hypothesis .
Standard threshold for autosomal linkage: 1000
Standard threshold for autosomal nonlinkage: 1/100
Reference: JO, Sec. 4.4
Let  be pedigree likelihood when disease is linked
be pedigree likelihood when disease is linked
Let  be pedigree likelihood when disease is unlinked
be pedigree likelihood when disease is unlinked
The lod score is 
Lod score of magnitude > z mathematically implies
the statistical probability of error is  ,
ignoring multiple testing issues.
,
ignoring multiple testing issues.
Caution: What LINKAGE/FASTLINK prints for log(L) may be off by an additive term, but lod scores are correct.
Why do linkage analysis experts talk about lod scores rather than likelihoods?
Logarithms convert multiplication/division into addition/subtraction. This is very useful for multiple data sets.



So, the lod score can be computed as :

Use 150-400 polymorphic probes at 8--20cM spacing.
Always do two locus analysis between disease and marker probe of known position.
Interested in lodscores above 1 or 2.
Standard marker sets suggested by CHLC.
Two-stage scans can reduce lab costs.
Never, never compare in print the lod score with marker A against the lodscore with marker B
Use multiple probes at 1--4cM spacing.
Do two or three locus analysis between disease and marker probes of known position.
Interested in lodscores above 3.
Can find suitable markers from a combination of the Marshfield, MIT, and Généthon maps.
Use markers whose relative order is agreed upon.
Fixed marker map: 
Find best vector for each of 4 placements of
disease D.
Let the best likelihood for each order
be , 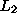 ,
,  ,
, .
.
Compare differences between log(), log(), log(), log()
If  then the odds that
j is incorrect in favor of i, are:
then the odds that
j is incorrect in favor of i, are: 
Desired level for t: 3.0
May have very high lodscores, yet uncertain marker order
Published maps may be incorrect over small regions
Published values are estimated from small samples
Limited variation among samples
Association between marker alleles and disease phenotypes (linkage disequilibrium) can confound the computations
Differ in: How thetas are chosen, output format
MLINK: Used to vary 1 at a time
LINKMAP: Used to vary 1 or 2 adjacent
at a time
ILINK: Used to find a locally optimal vector.
Locally optimal means that it cannot be perturbed by a small amount to get a different vector with a better likelihood.
VITESSE coalesces LINKMAP and MLINK by taking the LINKAGE program name as a parameter.
You want a table of 2-point lodscores comparing disease against a variety of markers and for a variety of theta values.
You need to fill in the lodscore at specific theta vectors.
Used for genome scan.
Caution: Avoid publishing positive results based solely on 2-point MLINK analysis. This error is common in the literature. It used to be excusable due to running time limitations.
You want to draw a multipoint lodscore plot.
You want to find in which gap of a fixed marker order the disease gene lies.
Major Caution: LINKMAP gives relatively little information about whether the disease may be near but outside the set of fixed markers.
Note: LINKMAP will not draw anything, but it will yield a table of values that can be plotted.
Whenever you have time to wait
When you do not feel the published thetas are valid for your data set
When you wish to estimate allele frequencies (too advanced for this course)
In the context of GenoCheck to find genotyping errors.
Major Caution: The output of ILINK is hard to understand. See README.ILINK in FD.
ILINK avoids reliance on published distances and may lead to stronger results.
Suppose the fixed map is
Suppose we established linkage between D and this region.
Suppose that there are  and
and  recombinants.
recombinants.
Conclude that D is between and .
This method of inference is incomparable in power to the likelihood method.
Published by CEPH/Généthon for many markers using a reference panel of families. See list of FTP/WWW sites.
Results are quite sensitive to allele frequencies
Allele frequencies can be estimated from input data.
Caution: All families in reference panel are from France and Utah.
Reference: TO, Chapter 22; Jim Tomlin's allele frequency estimation recipe
Dominant, Recessive, Co-dominant
Notation:
Dd: Heterozygous for disease
dd: Homozygous for non-disease
Penetrance, f: Probability that an individual of a given genotype is affected
In practice you may know affection status, but not genotype
Dominant:
Recessive:
Pure Codominant:
Must See: TO, Chapter 9
makeped: converts initial pedigree file into properly formatted file
preplink: interactively defines locus descriptions
lcp: prepares a script for execution of a sequence of main programs
lsp: used within script to extract desired loci from pedigree file
unknown: infers missing genotypes
lrp: converts the results of LINKMAP into a nice table suitable for multipoint graphing
``Nous vous conseillons de surveiller votre mail pendant les quelques premieres minutes apres le lancement. En effet si pour une raison quelconque (et elles sont nombreuses!) votre calcul `crashe' vous n'attendrez pas inutilement avant d'y remedier.''-Lucien Bachner (Infobiogen, Paris)
Reference: FD: README.trouble
PEDCHECK is a useful program to identify "inconsistent" genotypes. E.g., a child has allele 4, but neither parent does.
Each person has two alleles at each locus.
A child inherits one allele from the father and one allele from the mother.
The set of sibling alleles S can be written so that

in such a way that M and F are of size 1 or 2 and each sibling has 1 allele in M and 1 allele in F.
Most linkage programs check these rules.
A female has two alleles at a locus.
A male has one allele at a locus.
A female inherits her father's allele and one of her mother's alleles.
A male inherits one of his mother's alleles
The set of sibling alleles S can be written so that:
in such a way that M is of size 1, F is of size 1 or 2, each male sibling has 1 allele in F, each female sibling has 1 allele in M and 1 allele in F.
Genotypes can be wrong, but still consistent with Mendelian rules.
Wrong genotypes usually increase the estimate of .
One can estimate between markers using ILINK.
Estimated >> published is a good clue that there
are errors.
Use GenoCheck (derived from ILINK) to identify the most suspect person/locus pairs.
Convince laboratory personnel to follow-up based on GenoCheck output.
Reference: CD
On UNIX: Use ps (with flags) and look for a process running one of: ilink, linkmap, mlink.
Make sure that process status is R
Each likelihood evaluation takes roughly the same amount of time.
Programs take a checkpoint after every likelihood evaluation.
Use: ls -lt check*
to see when last checkpoint was taken.
Use parental pairs number
Reference: FD: README.time
Affected relatives have disease for the same reason
Basic method is affected sibpairs
Newer software considers affected relative pairs
Useful when mode of inheritance is unclear
APM, GENEHUNTER, SimIBD
Identical By State vs. Identical By Descent
Reference: TO, Chapter 26
 = probability that i,j share an allele IBS
= probability that i,j share an allele IBS
A = Sum of over all affected i,j pairs
Can also weight A by allele frequency.
Compare observed A to expected.
Very sensitive to allele frequencies.
Empirical p-values.
General approach using inheritance vectors.
Inheritance vector: each allele is from grandpa/grandma?
Given a probability distribution of inheritance vectors, conditional on phenotypes, compare this to uniform distribution.
Two different statistics:  ,
, 
somewhat analogous to A, but using IBD.
Obtain a normally distributed Z-statistic for p-values.
Reference: GD
A different statistic: SimIBD
Improves APM by conditioning simulation on affection status.
Improves APM by moving away from IBS to IBD.
You get empirical p-values.
Currently does only 2-point analysis
Reference: SD
A run of a program is an experiment.
An effective bug report enables the software developer to exactly reproduce the experiment without bias as to the outcome.
Use e-mail, not phone or FAX, but beware that mailers can mangle files.
Send files as ASCII, not BinHex
State what problems occurred, but describe only effects (phenotypes) that you observe, not causes (genotypes) that you conjecture.
Reference: FD: README.bugreport
Look for:
Look for:
These 7 items are not sufficient, but ought to be necessary for a "parametric" analysis. If any of the 7 items is missing, do not recommend the manuscript be accepted.
ftp: login as anonymous, leave full e-mail as password
http://bimas.cit.nih.gov/linkage/index.html
http://bimas.cit.nih.gov/linkage/galaxy1.html
http://linkage.rockefeller.edu
ftp watson.hgen.pitt.edu
under pub/apm
ftp fastlink.nih.gov
under pub/fastlink
http://waldo.wi.mit.edu/ftp/distribution/software/g enehunter/gh2
ftp softlib.cs.rice.edu
under pub/GenoCheck
ftp linkage.rockefeller.edu
under software/linkage/DOS
under software/utilities
under software/linkage/UNIX
and other subdirectories also
ftp watson.hgen.pitt.edu
under pub/pedcheck
ftp wastson.hgen.pitt.edu
under pub/simibd
ftp watson.hgen.pitt.edu
under pub/vitesse
ftp://www.genethon.fr/pub/Gmap/Nature-1995
http://research.marshfieldclinic.org/genetics
Follow various links to find markers or build maps.
http://www.genome.wi.mit.ediClick on Human Physical Mapping Project