| Cellular and Molecular Neurosciences Section | ||
| Mark P. Mattson, Ph.D., Chief Senior Investigator | ||
| Overview: The Cellular and Molecular Neurosciences Section employs a multifaceted array of experimental models of aging and age-related neurodegenerative disorders in order to establish the molecular and biochemical changes that occur during aging and in disorders such as Alzheimer's, Parkinson's and Huntington's diseases and stroke. Data obtained in these experimental models are integrated with data obtained in studies of both normal elderly humans and patients with neurodegenerative disorders to arrive at conclusions as to why neuronal dysfunction and degeneration occur in the disorders. In addition to identifying the molecular and cellular alterations that lead to neuronal degeneration in age-related neurological disorders, investigators are elucidating the cellular signaling mechanisms that allow successful brain aging. | ||
| In general, the projects underway fall into the following areas: | ||
| Ageing and neuronal vulnerability
Mark P. Mattson and Tim Magnus Everyone ages, but only some will develop a neurodegenerative disorder in the process. Disease might occur when cells fail to respond adaptively to age-related increases in oxidative, metabolic and ionic stress, thereby resulting in the accumulation of damaged proteins, DNA and membranes. Determinants of neuronal vulnerability might include cell size and location, metabolism of disease-specific proteins and a repertoire of signal transduction pathways and stress resistance mechanisms. Emerging evidence on protein interaction networks that monitor and respond to the normal ageing process suggests that successful neural ageing is possible for most people, but also cautions that cures for neurodegenerative disorders are unlikely in the near future. Click Here for PDF copy of this paper |
||
|
|
 |
|
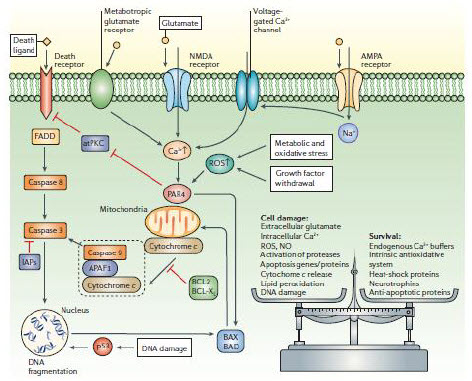
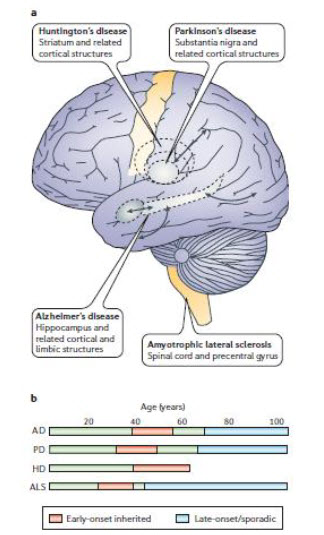
| Figure 1. The who, where and when of neuronal death in age-related neurodegenerative disorders. |
Figure 4. Once triggered, the death of neurons is programmed. |
|
|
||
Best in Small Doses Sometimes a smidgen of toxin can be just what the doctor ordered, Click Here for a PDF copy of this article
|
|
|
| Pathways towards and away from Alzheimer’s disease
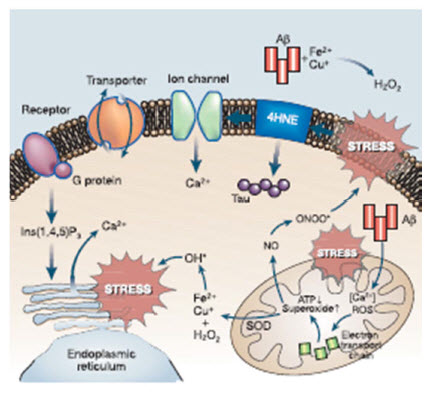
Mark P. Mattson - Laboratory of Neurosciences, National Institute on Aging Intramural Research Program, 5600 Nathan Shock Drive, Baltimore, Maryland 21224, and Department of Neuroscience, Johns Hopkins University School of Medicine, 725 N. Wolfe Street, Baltimore, Maryland 21205, USA Slowly but surely, Alzheimer’s disease (AD) patients lose their memory and their cognitive abilities, and even their personalities may change dramatically. These changes are due to the progressive dysfunction and death of nerve cells that are responsible for the storage and processing of information. Although drugs can temporarily improve memory, at present there are no treatments that can stop or reverse the inexorable neurodegenerative process. But rapid progress towards understanding the cellular and molecular alterations that are responsible for the neuron’s demise may soon help in developing effective preventative and therapeutic strategies. Click Here for a PDF copy of this article |
||
 |
 |
|
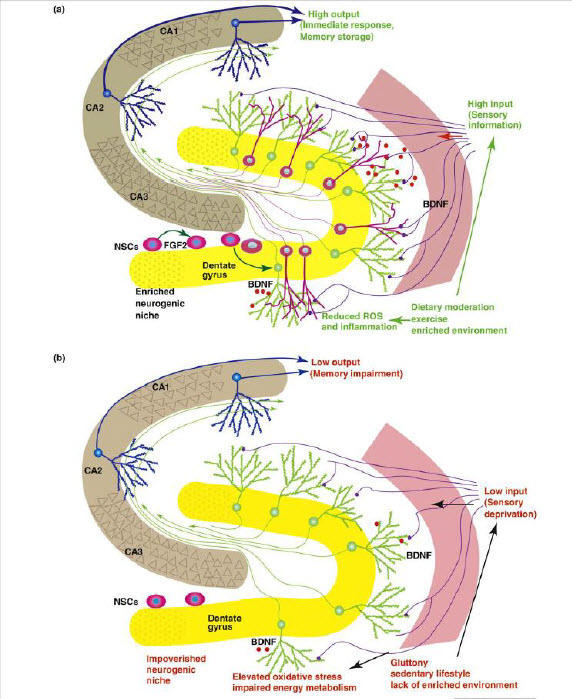
When neurogenesis encounters aging and disease. Lazarov O, Mattson MP, Peterson DA, Pimplikar SW, van Praag H - Department of Anatomy and Cell Biology, University of Illinois at Chicago, Chicago, IL, USA. In this review, we consider the evidence that a reduction in neurogenesis underlies aging-related cognitive deficits and impairments in disorders such as Alzheimer's disease (AD). The molecular and cellular alterations associated with impaired neurogenesis in the aging brain are discussed. Dysfunction of presenilin-1, misprocessing of amyloid precursor protein and toxic effects of hyperphosphorylated tau and β-amyloid probably contribute to impaired neurogenesis in AD. Because factors such as exercise, environmental enrichment and dietary energy restriction enhance neurogenesis, and protect against age-related cognitive decline and AD, knowledge of the underlying neurogenic signaling pathways could lead to novel therapeutic strategies for preserving brain function. In addition, manipulation of endogenous neural stem cells and stem cell transplantation, as stand-alone or adjunct treatments, seems promising. Click Here for a PDF copy of this article |
|
|
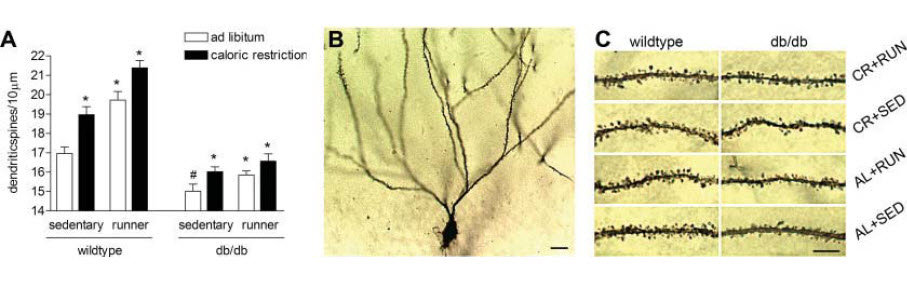
Voluntary Exercise and Caloric Restriction Enhance Hippocampal Stranahan AM, Lee K, Martin B, Maudsley S, Golden E, Cutler RG, Mattson MP - Psychology department, Princeton University, Princeton, New Jersey, USA. Erratum in: Hippocampus. 2009 Nov;19(11):1151. Diabetes may adversely affect cognitive function, but the underlying mechanisms are unknown. To investigate whether manipulations that enhance neurotrophin levels will also restore neuronal structure and function in diabetes, we examined the effects of wheel running and dietary energy restriction on hippocampal neuron morphology and brain-derived neurotrophic factor (BDNF) levels in db/db mice, a model of insulin resistant diabetes. Running wheel activity, caloric restriction, or the combination of the two treatments increased levels of BDNF in the hippocampus of db/db mice. Enhancement of hippocampal BDNF was accompanied by increases in dendritic spine density on the secondary and tertiary dendrites of dentate granule neurons. These studies suggest that diabetes exerts detrimental effects on hippocampal structure, and that this state can be attenuated by increasing energy expenditure and decreasing energy intake. Click Here for a PDF copy of this article |
||
|
||
Disrupted energy metabolism and neuronal circuit dysfunction in cognitive impairment and Alzheimer’s disease Dimitrios Kapogiannis, Mark P Mattson Epidemiological, neuropathological, and functional neuroimaging evidence implicates global and regional disruptions in brain metabolism and energetics in the pathogenesis of cognitive impairment. Nerve cell microcircuits are modifi ed by excitatory and inhibitory synaptic activity and neurotrophic factors. Ageing and Alzheimer’s disease cause perturbations in cellular energy metabolism, level of excitation or inhibition, and neurotrophic factor release, which overwhelm compensatory mechanisms and result in dysfunction of neuronal microcircuits and brain networks. A prolonged positive energy balance impairs the ability of neurons to adapt to oxidative and metabolic stress. Results from experimental studies in animals show how disruptions caused by chronic positive energy balance, such as diabetes, lead to accelerated cognitive ageing and Alzheimer’s disease. Therapeutic interventions to allay cognitive dysfunction that target energy metabolism and adaptive stress responses (such as neurotrophin signalling) have been eff ective in animal models and in preliminary studies in humans. Click Here for a PDF copy of this article |
||
 |
 |
|
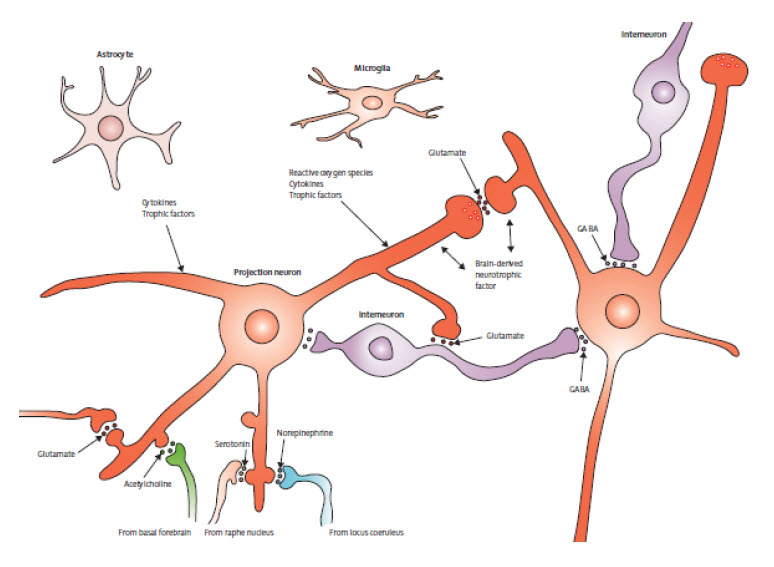
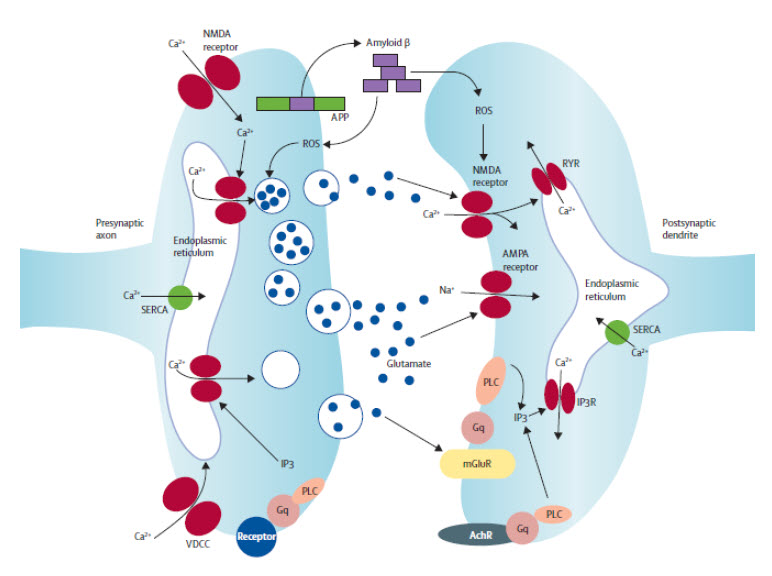
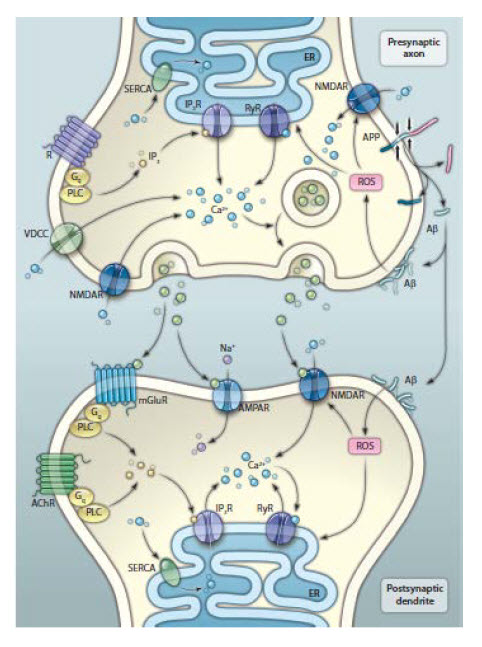
| Figure 3: Basic organisation of neuronal microcircuits involved in cognitive processing The major excitatory projection neurons are glutamatergic and their axons synapse onto dendrites of other glutamatergic neurons that might, in turn, project their axons to a diff erent brain region. GABAergic interneurons receive excitatory inputs from glutamatergic neurons and form synapses on the cell bodies of the same or other glutamatergic neurons. Glutamatergic neurons also receive synaptic inputs from neurons modulated by norepinephrine, serotonin, and acetylcholine. The cell bodies of these neurons are located in the locus coeruleus, raphe nucleus, and basal forebrain, respectively. Neurons in all brain regions also interact with glial cells including astrocytes and microglia, which produce trophic factors and cytokines that might normally be important in synaptic plasticity. However, excessive production of proinfl ammatory cytokines and reactive oxygen species by glial cells has been implicated in the pathogenesis of cognitive impairment and Alzheimer’s disease. | Figure 4: Mechanisms of synaptic dysfunction in ageing and Alzheimer’s disease APP is axonally transported and so is present in high concentrations in presynaptic terminals. In properly functioning synapses, APP is proteolytically cleaved in the middle of the amyloid β sequence by α secretase, thereby preventing the production of amyloid β. During normal ageing, and more so in Alzheimer’s disease, APP is cleaved at the N-terminal and C-terminal of amyloid β by β secretase and γ secretase, respectively, resulting in the production and self-aggregation of amyloid β. Aggregation of amyloid β on the membrane generates ROS, which results in membrane lipid peroxidation. This in turn impairs the function of membrane ion-motive ATPases, thereby promoting membrane depolarisation and calcium infl ux through NMDA receptor channels and voltage-dependent calcium channels. Sustained increases in cytoplasmic calcium concentrations promote depletion of presynaptic glutamate stores, resulting in impaired synaptic transmission and damage to axons and dendrites. Additionally, disrupted mitochondrial function caused by ageing, oxidative stress, and amyloid β results in energy depletion in neurons, which exacerbates synaptic dysfunction and degeneration of neurons. Dysregulation of endoplasmic reticulum function, which results in depletion of endoplasmic reticulum calcium stores and accumulation of misfolded proteins further contributes to the death of neurons in Alzheimer’s disease. APP=amyloid precursor protein. ROS=reactive oxygen species. SERCA=sarco/endoplasmic reticulum Ca²+-ATPase. RyR=ryanodine receptor. IP3R=inositol trisphosphate receptor. PLC=phospholipase C. mGluR=metabotropic glutamate receptor. AchR=acetylcholine receptor. VDCC=voltage-dependent calcium channel. | |
ER Calcium and Alzheimer’s Disease: Mark P. Mattson - Published 23 March 2010; Volume 3 Issue 114 pe10
Click Here for a PDF copy of this article
|
|
|
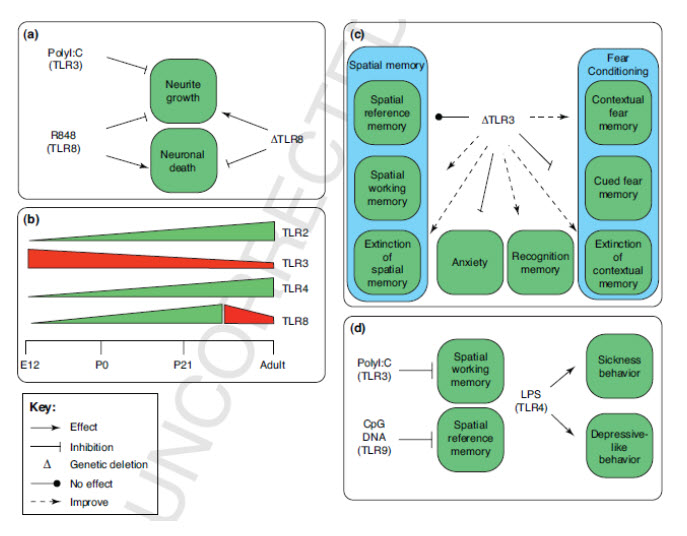
Toll-like receptor Signaling in Neural Plasticity and Disease Eitan Okun, Kathleen J. Griffioen and Mark P. Mattson - Laboratory of Neurosciences, National Institute on Aging Intramural Research Program, Biomedical Research Center, Baltimore, MD Abstract Toll-like receptors (TLRs) are a family of innate immune system receptors that respond to pathogen-derived and tissue damage-related ligands. TLR signaling in immune cells, glia and neurons may play roles in the pathogenesis of stroke, Alzheimer’s disease and multiple sclerosis. Recent findings suggest that TLR signaling also influences multiple dynamic processes in the developing and adult central nervous system including neurogenesis, axonal growth and structural plasticity. In addition, TLRs are implicated in the regulation of behaviors including learning and memory, and anxiety. This review describes recently discovered and unexpected roles for TLRs in neuroplasticity, and the implications of these findings for future basic and translational research studies. Click Here for a PDF copy of this paper (Final Accepted Manuscript) |
||
 |
||

|
Help Downloading Files on This Page IRP Home What's New Contact Us Accessibility Disclaimer Privacy Site Map NIA Home |
|
|