New Targeted Therapies Available for Licensing: Inhibitors of Polo-like Kinase 1
While a Postdoctoral Fellow at Harvard University studying cellular proliferation and mitotic controls, Kyung Lee, Ph.D., made a seminal discovery of the key role played by the noncatalytic polo-box domain (PBD) of mammalian polo-like kinase 1 (Plk1). He demonstrated that the PBD plays an essential role for the function of Plk1 by targeting its catalytic domain to specific subcellular structures. Plk1 is of interest to cancer research because it is over-expressed in a broad spectrum of human cancers, and also because blockade of Plk1 function efficiently kills cancer cells, but much less efficiently kills normal cells, due to the addiction of the former to a high level of Plk1 activity.

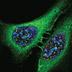
A high affinity PBD inhibitor, 4j (yellow ball and stick) disrupts Plk1 (red fluorescent signals in the immunostained cells; green, alpha-tubulin; blue, chromosome) localization and induces cancer cell killing. Binding by 4j to the PBD is achieved by the rotation (curved arrow) the Y481 side chain (semi-transparent cyan), which reveals a newly discovered hydrophobic channel on the surface of the Plk1 PBD. (T. Burke and K. Lee, CCR)
After coming to CCR in 1998 as a tenure-track investigator in the Laboratory of Metabolism, Lee discovered a 5-residue long phospho-peptide, PLHSpT, which specifically binds to the Plk1 PBD with a high affinity. He sought the expertise of CCR investigators, Alexander Wlodawer, Ph.D., Chief of the Macromolecular Crystallography Laboratory, and James McMahon, Ph.D., Chief of the Molecular Targets Laboratory, to determine the exact crystal structure and binding mode of the PBD-PLHSpT complex.
This collaboration provided Lee’s research team with the molecular look they needed to better understand the nature of Plk1 PBD-dependent interactions. Lee and his colleagues further found that disrupting the function of Plk1 PBD by PLHSpT derivatives induces mitotic arrest and apoptotic cell death in cultured cancer cells. Given that catalytic domain inhibitors frequently exhibit a high level of cross-reactivities, Lee suspected that Plk1 PBD inhibitors might make excellent targeted therapies for cancer because of their unparalleled affinity and specificity. Consequently, in the spring of 2008, Lee approached Terrence Burke, Ph.D., in CCR’s Chemical Biology Laboratory, to explore the possibility of further developing the PLHSpT-based inhibitors. Since that meeting, Burke’s expertise in the generation of peptide derivatives coupled with the biochemical and cell-based analyses of Plk1-dependent events performed by Lee’s lab, has yielded several highly potent Plk1 PBD-binding compounds.
As Lee explains, “While systematically analyzing various PBD-binding proteins, my research team and I identified a PLHSpT-containing motif from a kinetochore protein. Next, synthetic work in Burke’s laboratory led to the serendipitous discovery that certain modifications to the natural amino acid histidine within the PLHSpT peptide imparts up to a 1000-fold better binding affinity to PBD.”
“This,” according to Burke, “makes CCR’s Plk1 inhibitors currently the most potent ones yet reported, because we managed to maintain good Plk1 target selectivity along with exceptional binding affinity.”
The Plk1 PBD-binding inhibitors are currently in preclinical development and are available for licensing through the NIH Office of Technology Transfer.
To inquire about licensing Plk1 PBD-binding inhibitors, please contact Patrick McCue, Ph.D., at McCuepat@mail.nih.gov.
For a description of the new agents, please visit http://www.ott.nih.gov/Technologies/
abstractDetails.aspx?RefNo=1971.
To learn more about Dr. Lee’s research, please visit his CCR Web site at
http://ccr.cancer.gov/staff/staff.asp?name=leeks.
To learn more about Dr. Burke’s research, please visit his CCR Web Site at http://ccr.cancer.gov/staff/staff.asp?Name=burke.
News Articles


Smurf2 Regulates DNA Repair and Packaging to Prevent Tumors
The blueprint for a cell’s functions is written in the genetic code…

Finding a Chink in the Armor: Investigating the Structure of HIV
HIV infection depends on two proteins expressed on the virus surface: gp41…

Recent CCR Awards
View CCR Awards…

Staff News at CCR
Staff announcements at CCR…

CTCF, a Novel Regulator of Alternative Splicing
CTCF binding may play an important role in exon 5’s inclusion in the CD45 mRNA transcript…

In Conversation: Research Fellow Sanjeev Shukla, Ph.D.
Among the multiple alterations present in cancer cells are an abundance of aberrant mRNA transcripts…

Collaborating to Outwit
HLRCC’s Pathways
Marston Linehan, M.D., has spent his entire career at CCR crafting better approaches to treating kidney cancer…