
Lab Home
Head: Juan S. Bonifacino, Ph.D.
We study the molecular mechanisms involved in protein sorting in the endocytic and secretory pathways. The interior of the cell is organized into an array of membrane-bound compartments, each of which is composed of a specific set of resident proteins. The localization of transmembrane proteins to these compartments is mediated by short sequences of amino acids that function as sorting signals. These signals bind to receptor-like molecules (i.e., "adaptors") that connect the signals to the sorting machinery. We focus on two types of signal, referred to as tyrosine-based and dileucine-based sorting signals, which are involved in protein internalization from the cell surface, targeting to endosomes and lysosomes, delivery to the basolateral plasma membrane of polarized epithelial cells, and localization to specialized endosomal/lysosomal organelles such as melanosomes and platelet dense bodies.
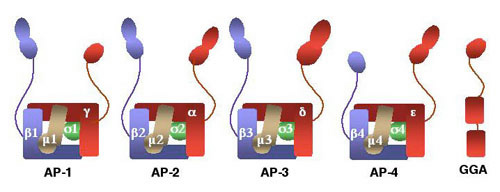
Years ago, we discovered that tyrosine-based sorting signals fitting the YXXØ tetrapeptide motif (Y is tyrosine, X is any amino acid, and Ø is a bulky hydrophobic amino acid) are recognized by the m1 and m2 subunits of the clathrin-associated adaptor complexes AP-1 and AP-2, respectively. In addition, we and others discovered two additional adaptor complexes, named AP-3 and AP-4. The m3 and m4 subunits of these complexes also recognize YXXØ signals. The figure below shows a schematic representation of the four AP complexes and their subunits.
 The AP-3 adaptor complex is widely expressed from yeast to humans. This has allowed genetic analyses of AP-3 function in model organisms. Studies in Drosophila demonstrated that altered expression of any of the four subunits of AP-3 (i.e., d, b 3, m3 or s3) results in a pigmentation defect. This is due to impaired biogenesis of pigment granules, which share a common biogenetic pathway with lysosomes. This work, together with findings from other laboratories in yeast and mice, indicate that AP-3 is involved in protein sorting to specialized endosomal-lysosomal organelles. In collaboration with Dr. William Gahl, we found that mutations in the b3A subunit isoform of AP-3 are the cause of Hermansky-Pudlak syndrome type 2, a pigmentation and bleeding disorder. We also contributed to the identification of multi-subunit complexes named BLOC (for biogenesis of lysosome-related organelles complex), which are defective in seven other types of the Hermansky-Pudlak syndrome.
The AP-3 adaptor complex is widely expressed from yeast to humans. This has allowed genetic analyses of AP-3 function in model organisms. Studies in Drosophila demonstrated that altered expression of any of the four subunits of AP-3 (i.e., d, b 3, m3 or s3) results in a pigmentation defect. This is due to impaired biogenesis of pigment granules, which share a common biogenetic pathway with lysosomes. This work, together with findings from other laboratories in yeast and mice, indicate that AP-3 is involved in protein sorting to specialized endosomal-lysosomal organelles. In collaboration with Dr. William Gahl, we found that mutations in the b3A subunit isoform of AP-3 are the cause of Hermansky-Pudlak syndrome type 2, a pigmentation and bleeding disorder. We also contributed to the identification of multi-subunit complexes named BLOC (for biogenesis of lysosome-related organelles complex), which are defective in seven other types of the Hermansky-Pudlak syndrome.
In addition, we described a new family of monomeric clathrin adaptors named GGAs. The GGAs have a modular structure consisting of VHS, GAT, hinge and GAE domains. Each of these domains binds a different set of ligands. The VHS domain binds DXXLL-type signals (D is aspartate, X is any amino acid, and L is leucine) present in the cytosolic domains of the mannose 6-phosphate receptors. The GAT domain binds the GTP-bound form of ARF and serves to recruit the GGAs to the trans-Golgi network. In addition, the GAT domain binds ubiquitin and thus plays a role in the sorting of ubiquitinated transmembrane proteins. The hinge domain binds clathrin. Finally, the GAE domain binds accessory factors such as rabaptin-5, enthoprotin, gamma-synergin, aftiphilin and p56. These interactions indicate that the GGAs function as ARF-dependent clathrin adaptors for the sorting of mannose 6-phosphate receptors and ubiquitinated transmembrane proteins at the trans-Golgi network.
Finally, we showed that another multi-subunit complex named retromer plays a role in the retrograde transport of mannose 6-phosphate receptors from endosomes to the trans-Golgi network.
Ongoing projects in the laboratory include the study of the structure, localization and role in protein sorting of adaptor proteins and their interaction partners. From these studies, we hope to contribute to a better understanding of the molecular mechanisms by which transmembrane proteins are delivered to different compartments of the endocytic and secretory pathways, and of the diseases that result from dysfunction of these mechanisms.


