

Grantee explores the relationship between DNA damage and aging
By John House

Niedernhofer’s talk offered a more detailed look at new findings she presented the previous day at the annual meeting of Outstanding New Environmental Scientist (ONES) awardees. Her research in aging has yielded important biomarkers of biological aging, given key insight into mechanisms of aging, and hinted at therapeutic approaches to delay the onset of aging. (Photo courtesy of Steve McCaw)

The talk drew a capacity crowd of NIEHS scientists, including some of the top researchers in the Laboratory of Molecular Genetics and Laboratory of Structural Biology. NIEHS Health Science Administrator Kimberly McAllister, Ph.D., was host for the talk. (Photo courtesy of Steve McCaw)
At first look, aging seems to be genetically programmed. Babies look the same and share common features that change in concert between individuals as they age, and the disparate life spans of different species clearly points to a strong genetic component. But a new way of looking at the genetic component in aging suggests the process may be much less programmed than shaped by a time-dependent accumulation of damage to mitochondria, lamins, DNA, RNA, and proteins, with an important part of the damage driven by environmental exposures.
Grantee Laura Niedernhofer, M.D., Ph.D., (http://www.cbp.pitt.edu/faculty/niedernhofer.html) finds her passion in attempting to answer the burning questions of what types of DNA damage cause aging and through what mechanisms. Her talk at NIEHS July12 presented recent work she has conducted with NIEHS support. (http://projectreporter.nih.gov/project_info_description.cfm?aid=8097589&icde=13181929&ddparam=&ddvalue=&ddsub=&cr=6&csb=default&cs=ASC) She expressed her gratitude for her grant by saying, “I’m just proud to be funded by NIEHS.”
Studying defects in DNA repair with a partial knockout model
Niedernhofer noted that many diseases associated with genome instability caused by a DNA repair defect, such ataxia telangiectasia (also known as Louis-Bar syndrome), Cockayne syndrome, and Werner syndrome, lead to premature aging or a cancer phenotype. Furthermore, approximately two percent of the genome is dedicated to repair of DNA damage, which makes sense given that humans are continually exposed to genotoxins from exogenous sources such as ultraviolet light and X-rays, as well as endogenous sources from metabolism and its production of radical oxygen species.
As Niedernhofer explained, work in the 1980s demonstrated the importance of the human DNA repair gene ERCC1 and subsequent work showed that ERCC1 forms a complex with XPF to assist with multiple types of DNA repair. Strikingly, mutations in XPF can cause premature aging or progeria, characterized by muscle wasting, osteoporosis, anemia, and neurodegeneration, among other degenerative changes.
Ercc1 knockout, (Ercc1(-/-)), mice exhibit severe neurodegeneration and dystonia and die within four weeks of birth. In order to study the role unrepaired DNA damage plays in aging, Niedernhofer created partial knockout (Ercc1(-/Δ)) mice that make around five percent as much of the ERCC1-XPF complex as wild-type animals. By the time these mice reach 25 weeks of age, they exhibit the physical characteristics seen in three-year-old wild-type mice and spontaneously develop cerebral atrophy, osteoporosis, thin skin, and other characteristics of the XPF progeroid syndrome, all from a defect in the ability to repair DNA damage.
Through what mechanism(s) does DNA damage promote aging?
Having shown ERCC1-XPF insufficiency in mice causes progeroid syndrome, as it does in humans, Niedernhofer is currently determining the mechanism(s) by which unrepaired DNA damage in Ercc1(-/Δ) mice drives premature aging. Her group determined oxidative DNA damage resulting in cyclopurine adducts was increased in tissues of Ercc1(-/Δ) mice compared to controls. To test the hypothesis that these DNA lesions were caused by mitochondrial-produced ROS, Ercc1(-/Δ) mice were chronically treated with a mitochondrial-targeted radical scavenger (XJB-5-131). Remarkably, this resulted in reduced cyclopurine adducts and delayed progeroid symptoms in the mice.
Different tissues responded differently to DNA repair deficiency. In Ercc1-(-/Δ)mice, neurons were particularly vulnerable to lack of Ercc1 and exhibited death; liver cells senesced, as shown by loss of regenerative ability in mice after partial hepatectomy, while the hematopoietic system suffered increased turnover and loss of the stem cell pool in BrdU labeling experiments.
Niedernhofer noted these responses pointed to cell autonomous responses to DNA damage left unrepaired in Ercc1(-/Δ) mice. To examine non-cell autonomous responses to DNA damage, she next turned her attention to nuclear factor-kappa beta (NF-kB). NF-kB is a transcription factor with multiple roles in cell signaling in response to damage.
Prior work by other groups had already implicated NF-kB in senescence and aging. NF-kB is upregulated in aged tissues and suppression of NF-kB in mouse reversed aging of skin. Nearly every tissue examined in Ercc1(-/Δ) mice had increased NF-kB activity. Moreover, inhibition of NF-kB in Ercc1(-/Δ) mice delayed onset of progeroid aging and reduced DNA damage. This and additional experiments suggest NF-kB signaling causes cellular responses that result in a feed-forward loop of cellular stress and additional DNA damage.
(John House, Ph.D., is a postdoctoral fellow in the NIEHS Lung Respiratory Biology Group.)
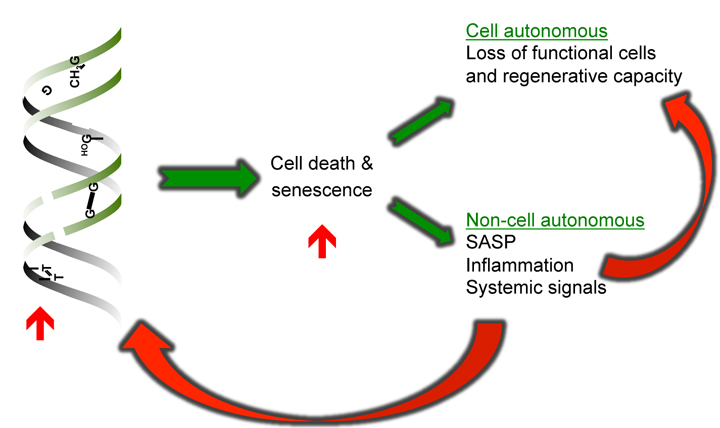
Mechanism by which stochastic damage drives aging
DNA damage, if not properly repaired, can result in cellular death and senescence through cell autonomous functions, including loss of functional cells and regenerative capacity as well as non-cell autonomous functions, such as paracrine signaling, senescence-associated secretory phenotype, and other systemic signals. These can result in a feed-forward loop that causes more DNA damage.
"Study examines the role ..." - previous story ![]()
![]() next story - "NIEHS summer interns learn ..."
next story - "NIEHS summer interns learn ..."
August 2012 Cover Page